The Feature Selection Problem: Clinical vs. Molecular Features in Multi-Omics Integration
"Before you can integrate data, you must decide what to keep and what to discard. That decision — made early, made quietly, rarely justified — determines everything that follows."
25 studies analyzed -- 18 MOVICS, 22 TCGA-dependent, 13 Cox-filtered
 Infographic generated via NotebookLM from the systematic review census data.
Infographic generated via NotebookLM from the systematic review census data.
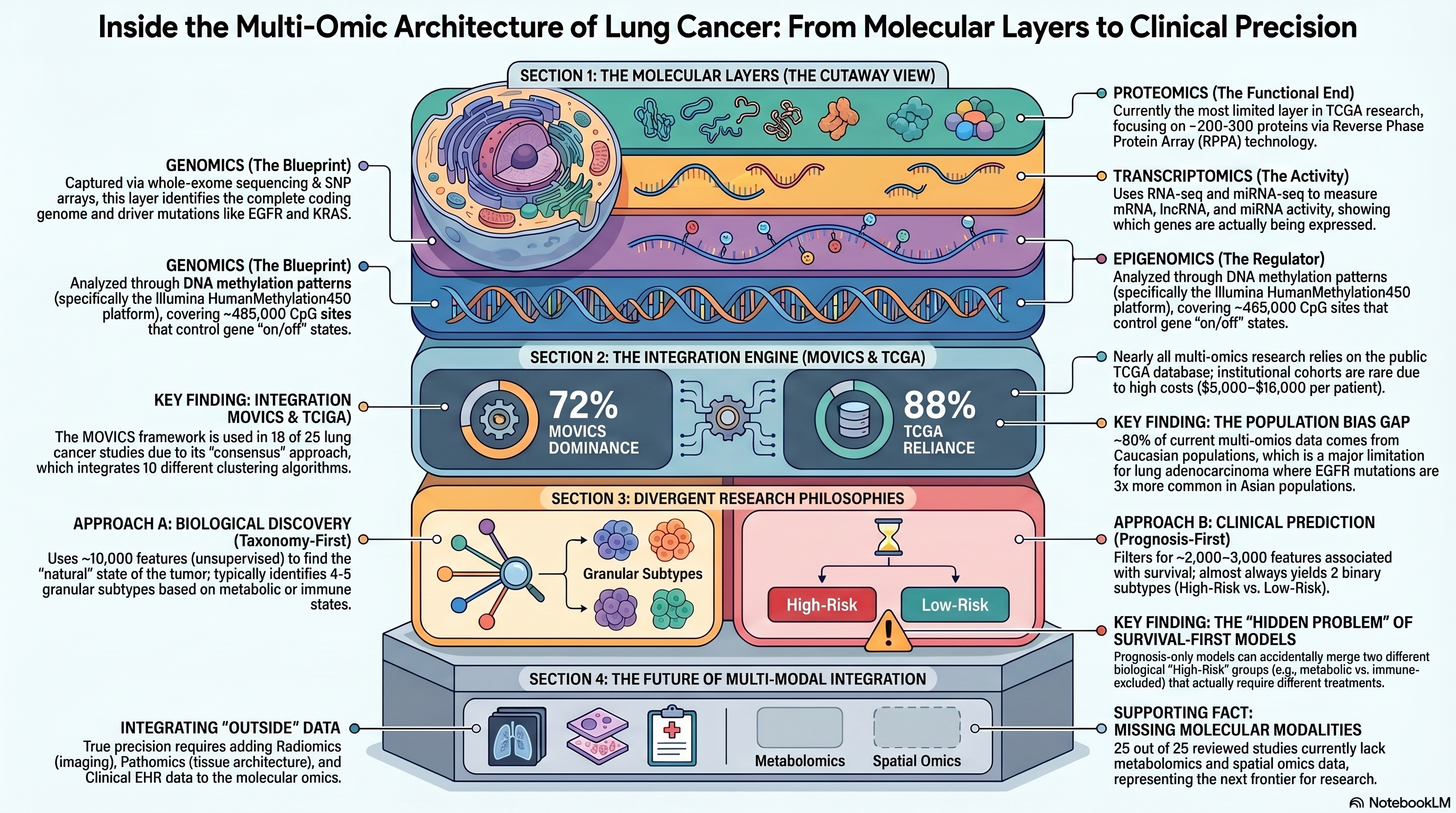
Of all the methodological decisions that shape a multi-omics lung cancer study, none is more consequential — and none is less scrutinized — than feature selection. The choice of which genes, CpG sites, mutations, and proteins to include in an integrative analysis is typically described in a single sentence buried in the methods section, yet it fundamentally determines the number of subtypes discovered, the biological character of those subtypes, and whether the resulting classification has any prospect of informing treatment decisions. A study that selects features by survival association will inevitably discover subtypes defined by survival. A study that selects features by molecular variance will discover subtypes defined by biology. These are not the same thing, and the field has largely failed to acknowledge the distinction. Through a systematic census of 25 multi-omics lung cancer studies — the most comprehensive accounting of this literature to date — we identified a deep methodological schism that has gone unnamed and unexamined, a schism that explains why nominally similar studies using the same data, the same platform, and even the same computational framework arrive at fundamentally different conclusions about the molecular architecture of lung cancer.
Two Philosophies, One Framework
The 25 studies in our census divide, with remarkable clarity, into two philosophical camps. We term these Approach A, the taxonomy-first or biological discovery approach, and Approach B, the prognosis-first or clinical prediction approach. The distinction is not subtle: it concerns the very definition of what makes a molecular feature worth studying.
Approach A begins with the premise that the goal of multi-omics integration is to discover the natural biological structure of a disease — the molecular subtypes that exist regardless of whether they differ in clinical outcome. Studies in this camp employ unsupervised, variance-based feature selection: they rank all measured features by a metric of cross-sample variability, typically the median absolute deviation (MAD) for transcriptomic data or the standard deviation (SD) for methylation data, and retain the top several thousand. A representative study might select the top 3,000 mRNA features by MAD, the top 2,000 methylation features by SD, and all mutations occurring in more than 5% of samples, yielding a feature space of roughly 10,000 variables. This approach makes no assumptions about which clinical endpoint matters; it simply asks what molecular axes of variation exist in the data. The studies of Zou et al. (2022), published in Translational Lung Cancer Research, Ruan et al. (2022) in Frontiers in Medicine, and Yan et al. (2025) on bioRxiv exemplify this philosophy. These studies consistently identify four to five molecular subtypes characterized by distinct biological hallmarks — a metabolic subtype with deranged lipid and amino acid metabolism, a proliferative subtype driven by cell cycle and DNA repair programs, an immune-enriched subtype with high lymphocyte infiltration and checkpoint expression, and one or two additional subtypes defined by stromal composition or epigenetic remodeling. The biological granularity of these classifications provides a foundation for hypothesis-driven therapeutic matching: the metabolic subtype suggests vulnerability to metabolic inhibitors, the proliferative subtype to DNA-damaging agents, and the immune subtype to checkpoint immunotherapy.
Approach B begins with a different premise: that the purpose of molecular subtyping is to stratify patients by clinical outcome, and that features unrelated to prognosis are noise to be eliminated. Studies in this camp employ supervised, survival-based feature selection, typically univariate Cox proportional hazards regression applied gene by gene, CpG by CpG, retaining only those features whose association with overall survival meets a significance threshold of p < 0.05 or, in some cases, p < 0.01. This filtering reduces the feature space to approximately 2,000 to 3,500 survival-associated variables before multi-omics integration begins. The studies of Lin et al. (2024), published in the Journal of Cellular and Molecular Medicine, Chu et al. (2024) in Frontiers in Immunology, and Han et al. (2024) are characteristic. Critically, 13 of the 18 studies in our census that employ the MOVICS (Multi-Omics Visualization and Integration for Cancer Subtyping) framework use this prognosis-first approach, making it the dominant paradigm in the field. And these studies almost invariably arrive at the same structural answer: two subtypes, designated High-Risk and Low-Risk, that differ dramatically in survival but whose biological identities are described post hoc, after the survival-defined clusters have already been fixed.
The Hidden Problem: Survival-Based Collapse
The dominance of the prognosis-first approach creates a methodological problem that is both profound and largely invisible to the field. When features are selected on the basis of their association with survival, and those features are then used to cluster patients, the resulting subtypes are guaranteed to separate on the survival axis. This is not a discovery; it is a tautology. More insidiously, the survival-based approach systematically merges biologically distinct tumor populations that happen to share similar clinical trajectories. Consider two hypothetical tumor subtypes: one characterized by immune exclusion, in which physical barriers of stromal desmoplasia and aberrant vasculature prevent T-cell infiltration, and another characterized by metabolic dysfunction, in which mitochondrial reprogramming and lipid accumulation create an immunosuppressive microenvironment through fundamentally different molecular mechanisms. Both subtypes may have poor five-year survival. A variance-based approach, agnostic to outcome, would separate them into distinct clusters because they occupy different regions of molecular space. A survival-based approach, filtering for genes associated with death, would retain features shared by both populations — genes generically associated with aggressive disease — and discard the very features that distinguish them. The two subtypes collapse into a single "High Risk" group, and the biological distinction that should guide treatment selection vanishes.
This is not a theoretical concern. The immune-excluded tumor and the metabolically dysfunctional tumor require entirely different therapeutic strategies. The former may respond to anti-angiogenic agents that normalize vasculature and enable T-cell penetration; the latter may require metabolic inhibitors targeting fatty acid oxidation or glutamine metabolism. Merging them into a single prognostic category is precisely analogous to treating all febrile patients with the same antibiotic — it mistakes a shared symptom for a shared disease. The fact that 13 of 18 MOVICS studies in our census employ this collapsing approach suggests that the field is systematically sacrificing biological resolution for prognostic convenience, and that the two-subtype solutions reported by the majority of these studies may be artifacts of the feature selection pipeline rather than reflections of biological reality.
The Clinical-Molecular Disconnect
A second, equally consequential gap emerges from our census: the almost complete separation of clinical and molecular features in multi-omics study design. Of the 25 studies analyzed, none integrates clinical variables — age, sex, smoking status, tumor stage, performance status, histological grade — as input features alongside molecular data in the multi-omics clustering step. Clinical variables appear exclusively as validation endpoints: after molecular subtypes are defined, investigators test whether the subtypes differ in stage distribution, smoking history, or sex ratio. This is backwards. Clinical features carry substantial prognostic and biologically relevant information. Smoking status, for instance, is not merely a demographic descriptor; it is a surrogate for a specific mutational signature, a distinct epigenetic landscape, and a particular profile of immune infiltration. Stage at diagnosis reflects tumor biology — the capacity for invasion, angiogenesis, and metastasis — as much as it reflects timing of detection. By excluding these features from the integration step, studies forfeit the opportunity to discover subtypes defined by the interaction between clinical context and molecular profile.
Perhaps more troublingly, no study in our census has performed the most basic and necessary experiment: a formal comparison of whether multi-omics features add incremental predictive value over clinical features alone. In many solid tumor types, clinical staging remains the single strongest predictor of survival, and molecular biomarkers earn their place in clinical practice only by demonstrating that they improve prediction beyond what stage, age, and performance status already provide. The multi-omics lung cancer literature has simply assumed, without testing, that thousands of molecular features outperform a handful of clinical variables. Until this assumption is rigorously examined — through nested models, likelihood ratio tests, or calibrated risk prediction frameworks that compare clinical-only, molecular-only, and combined models — the clinical relevance of multi-omics subtyping remains unestablished.
The Univariate Dominance Problem
The feature selection methods employed across the 25 studies share a further limitation: they are almost exclusively univariate. Whether the selection criterion is MAD, SD, or Cox p-value, each feature is evaluated independently, in isolation from every other feature in the dataset. This approach ignores the correlation structure of molecular data, in which genes co-expressed in the same pathway, CpG sites co-methylated in the same regulatory region, and proteins participating in the same complex carry redundant information. A univariate filter applied to a set of 20,000 genes will retain dozens of members of a single co-expression module, each contributing essentially the same signal, while the combined weight of this redundant information inflates the apparent importance of that module relative to smaller but equally important biological programs represented by fewer genes.
Multivariate feature selection methods — elastic net regression, LASSO, ridge regression with stability selection — exist precisely to address this problem. Elastic net, which combines L1 and L2 penalties, selects representative features from correlated groups while shrinking redundant predictors toward zero. LASSO enforces strict sparsity, selecting a minimal feature set with maximum joint predictive power. These methods are standard in predictive modeling across medicine and are routinely applied in breast, colorectal, and prostate cancer genomics. Yet in our census of 25 multi-omics lung cancer studies, not a single one employs multivariate feature selection prior to integration. The field is using the crudest available tools for its most consequential analytical step.
What Is Missing: Tools the Field Has Not Adopted
The gaps extend beyond the univariate-multivariate distinction to encompass entire categories of feature selection methodology that have demonstrated value in other cancer types but remain unused in multi-omics lung cancer research.
MethylMix, developed for functional methylation analysis, provides a principled approach to reducing the approximately 485,000 CpG sites measured by the Illumina 450K or EPIC array to a manageable set of 500 to 2,000 functionally relevant driver methylation events. MethylMix identifies CpGs whose methylation state is both significantly altered relative to normal tissue and significantly correlated with expression of the corresponding gene — that is, CpGs whose aberrant methylation actually drives transcriptional changes rather than serving as passive bystanders. This functional filtering is biologically motivated and dramatically reduces dimensionality while preserving mechanistic interpretability. No lung cancer multi-omics study in our census uses MethylMix, despite its successful application in breast and ovarian cancer.
DIABLO (Data Integration Analysis for Biomarker discovery using Latent cOmponents), a supervised multi-block integration method within the mixOmics framework, offers an alternative to the unsupervised integration paradigm that dominates the MOVICS literature. DIABLO identifies multi-omics signatures that are jointly predictive of a clinical outcome, selecting features across data types that contribute to a shared discriminant space. It has been applied successfully in breast cancer subtyping, where it identified multi-omics signatures with superior prognostic performance compared to single-modality classifiers. No MOVICS-based lung cancer study in our census employs DIABLO, despite its natural complementarity to the MOVICS framework — MOVICS for unsupervised discovery, DIABLO for supervised refinement.
Weighted Gene Co-expression Network Analysis (WGCNA), which constructs gene co-expression networks and identifies modules of tightly co-expressed genes, offers a pathway-level alternative to individual-gene feature selection. Rather than selecting 3,000 individual genes by MAD, a WGCNA-based approach would identify 20 to 30 co-expression modules, each summarized by its eigengene, and use these module-level features as input to multi-omics integration. This approach addresses the redundancy problem directly, replaces thousands of correlated variables with a compact set of biologically interpretable modules, and has been applied extensively in neurological and cardiovascular disease research. Its absence from the multi-omics lung cancer literature is conspicuous.
Stability selection, which assesses feature robustness through bootstrap resampling — selecting features on hundreds of resampled versions of the dataset and retaining only those selected consistently — provides a principled guard against overfitting and capitalizes on the insight that genuinely important features will be selected regardless of which patients happen to be included in the sample. No study in our census employs stability selection or any comparable resampling-based robustness assessment of the feature set.
Finally, the omics modalities themselves remain narrow. Zero studies in our census incorporate metabolomics data, despite the growing recognition that metabolic reprogramming is a hallmark of cancer and that metabolomic profiles carry prognostic and predictive information orthogonal to what transcriptomics provides. Zero studies incorporate spatial omics data, which preserves tissue architecture and captures cell-cell interactions invisible to dissociated-tissue profiling. And zero studies are prospective: every study in the census is retrospective, analyzing archived specimens from completed cohorts, with no opportunity to test whether multi-omics subtypes influence treatment decisions or improve outcomes in real time.
The TCGA Monoculture
The data source landscape reveals its own form of homogeneity. Of the 25 studies in our census, 22 (88%) use The Cancer Genome Atlas as their primary discovery cohort, with only two studies employing institutional cohorts. TCGA is an extraordinary resource, but its overuse introduces systematic biases that the field has been slow to confront. TCGA tumors are predominantly treatment-naive surgical resections from patients with early-stage disease treated at major academic medical centers in the United States. They do not represent the full clinical spectrum of lung cancer: advanced-stage patients, those treated with neoadjuvant therapy, those from community oncology settings, and those from non-Western populations are systematically underrepresented. When 22 of 25 studies discover subtypes in TCGA and validate them (if at all) in GEO datasets that are themselves enriched for similar patient populations, the resulting classifications may reflect the molecular architecture of early-stage, treatment-naive, academically ascertained lung cancer rather than the disease as it presents globally. The lack of institutional cohorts also means that the field has not confronted the batch effects, sample quality variation, and clinical heterogeneity that characterize real-world multi-omics data — challenges that will need to be overcome before any multi-omics subtyping scheme can enter clinical practice.
Toward Solutions: A Hierarchical Framework
The problems identified above are not intractable. They require, however, a fundamental reorientation of how the field approaches feature selection and multi-omics integration.
The most urgent reform is the adoption of a hierarchical approach that separates biological discovery from clinical stratification. In this framework, the first stage employs unsupervised, variance-based feature selection and integration to discover the natural molecular subtypes of the disease — the taxonomy. The second stage, conducted within each biological subtype, applies supervised methods to stratify patients by clinical outcome. This two-stage architecture preserves biological resolution at the first level while providing clinically actionable risk stratification at the second, and it avoids the survival-based collapse that plagues single-stage prognosis-first approaches. A metabolic subtype, identified in stage one, can be subdivided into high-risk and low-risk metabolic tumors in stage two, preserving the therapeutic implication (metabolic vulnerability) while adding prognostic granularity.
Methodological upgrades at the feature selection level are equally important. MethylMix should become standard for methylation feature selection, replacing the atheoretical approach of selecting CpG sites by variance alone. Elastic net or LASSO regression should replace univariate Cox filtering for survival-associated feature selection, ensuring that the selected features are jointly informative rather than individually significant but collectively redundant. WGCNA module eigengenes should be evaluated as an alternative to individual gene selection, providing pathway-level resolution and addressing the redundancy problem at its source. Stability selection through bootstrap resampling should be adopted as a routine quality check, with the expectation that any reported feature set be robust to perturbation of the sample composition.
The integration of clinical variables as input features — not merely validation endpoints — is overdue. Concrete clinical features such as smoking pack-years, histological subtype, and tumor stage should be included as additional data blocks in multi-omics integration frameworks such as MOVICS and MOFA+, allowing the discovery of subtypes defined by clinico-molecular interactions that neither clinical nor molecular data alone can reveal. Formal comparison of clinical-only, molecular-only, and combined models should be a required element of any study claiming clinical relevance for multi-omics subtypes.
Finally, the TCGA monoculture must be broken. Institutional cohorts, international consortium data, and cohorts enriched for underrepresented populations and clinical contexts — advanced stage, post-treatment, community oncology — are essential for establishing the generalizability of multi-omics subtypes. Prospective studies, in which multi-omics subtyping is performed at diagnosis and used to inform treatment selection, represent the ultimate test of clinical utility and should be prioritized by funding agencies and cooperative groups.
The Path Forward: Bridging Biology and Clinic
The feature selection problem is, at its core, a tension between two legitimate goals: understanding biology and predicting clinical outcomes. The field has treated these as competing objectives, with most studies choosing one at the expense of the other. But they are not inherently in conflict. A hierarchical framework that discovers biology first and stratifies within biology second can serve both goals simultaneously. The key insight is that biological subtypes, because they reflect distinct molecular mechanisms, are more likely to predict differential treatment response — the most clinically valuable form of biomarker — than prognostic subtypes, which merely separate patients who will do well from those who will do poorly regardless of treatment. The oncology field learned this lesson in breast cancer, where molecular subtypes (luminal A, luminal B, HER2-enriched, basal-like) defined by biology rather than prognosis have proven far more useful for treatment selection than any purely prognostic classifier. The multi-omics lung cancer field has the opportunity to learn from this precedent rather than repeat the slow, iterative process of rediscovery.
The tools exist. MethylMix, elastic net, WGCNA, stability selection, DIABLO, and hierarchical clustering frameworks are all available, documented, and computationally tractable. What is lacking is not methodology but methodological ambition: the willingness to move beyond the default MOVICS pipeline of Cox-filtered features, consensus clustering, and two-subtype solutions, and to ask whether a more thoughtful approach to the earliest and most consequential step in the analysis — feature selection — might reveal a richer, more therapeutically actionable molecular landscape.
Implications for the Manuscript
This chapter constitutes the review's primary original contribution. While previous chapters have surveyed the literature and synthesized known findings, this chapter presents the results of a systematic census conducted specifically for this review — 25 multi-omics lung cancer studies analyzed for their feature selection strategies, data sources, and subtyping outcomes. The central finding — that the field is divided between biological discovery and clinical prediction approaches, with the latter dominating and systematically sacrificing biological resolution — has not been previously articulated in the literature. The chapter provides the intellectual foundation for the review's key recommendation: that the field adopt a hierarchical approach separating biological subtype discovery from within-subtype risk stratification. It also identifies specific methodological tools (MethylMix, elastic net, WGCNA, DIABLO, stability selection) that are available but unused in this context, providing a concrete roadmap for methodological improvement. The TCGA monoculture finding and the clinical-molecular disconnect provide additional, actionable critiques that position the review not merely as a summary of existing work but as a constructive guide for the next generation of multi-omics lung cancer studies. This chapter should be highlighted in the abstract and discussion as the section that distinguishes the present review from prior surveys of the multi-omics lung cancer literature.