Theme 12 — Emerging Frontiers: Single-Cell, Spatial, and Multi-Modal Technologies
"Resolution is not merely a technical parameter — it is the difference between seeing a forest and understanding an ecosystem."
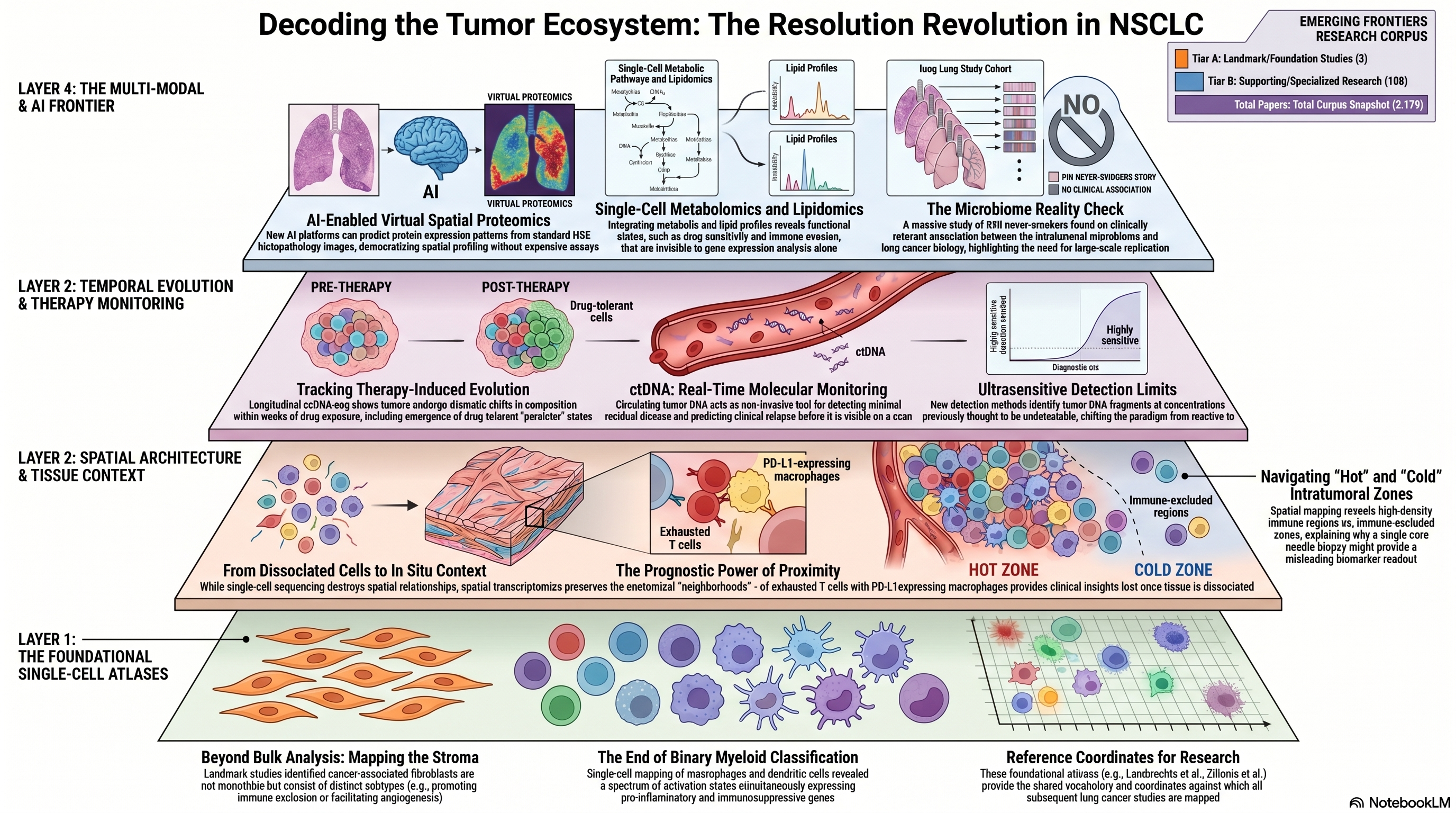
Corpus snapshot: 6,179 papers · 2 Tier A · 108 Tier B
The Resolution Revolution
The past half-decade has witnessed a transformation in the resolution at which lung cancer biology can be interrogated. Technologies that were exotic and expensive in 2018 — single-cell RNA sequencing (scRNA-seq), spatial transcriptomics, single-cell multi-omics — have matured into workhorse platforms generating datasets of unprecedented scale and dimensionality. This theme captures the frontier of that technological revolution as it intersects with NSCLC research: the dissection of tumor microenvironment heterogeneity at single-cell resolution, the mapping of spatial organization within intact tissue, and the integration of multiple molecular modalities to construct holistic portraits of tumor biology. With 2 Tier A papers and 108 Tier B publications, this theme bridges the gap between foundational single-cell atlasing efforts and the emerging wave of spatial and multi-modal studies that promise to reshape both basic understanding and clinical application.
Foundational Single-Cell Atlases of the NSCLC Microenvironment
The systematic deconstruction of NSCLC tumors into their cellular constituents began with landmark single-cell studies that revealed complexity invisible to bulk transcriptomic analysis. Lambrechts and colleagues produced one of the seminal scRNA-seq studies of the NSCLC stromal microenvironment, characterizing fibroblast, endothelial, and immune cell populations at single-cell resolution and identifying phenotypically distinct subpopulations within each lineage [PMID: 29988129]. Their analysis, cited over 1,350 times, demonstrated that cancer-associated fibroblasts are not a monolithic population but comprise functionally distinct subtypes — some promoting immune exclusion, others facilitating angiogenesis — with differential prognostic significance. This heterogeneity within the stromal compartment had been hypothesized but never directly observed at the resolution afforded by single-cell profiling, and its documentation fundamentally changed how the field conceptualizes the tumor microenvironment.
Complementing the stromal atlas, Zilionis and colleagues focused on the myeloid compartment, mapping the diversity of macrophage, monocyte, and dendritic cell populations within NSCLC tumors and adjacent normal lung tissue [PMID: 30979687]. Their work revealed a spectrum of myeloid activation states that defy simple M1/M2 classification, including tumor-specific macrophage programs characterized by simultaneous expression of pro-inflammatory and immunosuppressive genes. The functional ambiguity of these myeloid populations — neither cleanly pro-tumorigenic nor anti-tumorigenic — underscores the inadequacy of binary classification schemes and argues for continuous, rather than categorical, phenotypic frameworks. These foundational atlases established the reference coordinates against which subsequent studies have been mapped, creating a shared vocabulary for describing cellular heterogeneity in lung cancer.
Therapy-Induced Evolution at Single-Cell Resolution
The static view of tumor composition provided by single-timepoint atlases was extended into the temporal dimension by studies tracking how tumors evolve under therapeutic pressure. Maynard and colleagues performed a landmark longitudinal analysis using scRNA-seq to profile NSCLC tumors before, during, and after treatment with targeted therapy, demonstrating that drug exposure induces dramatic shifts in both tumor cell states and microenvironment composition [PMID: 32822576]. Their study documented the emergence of drug-tolerant persister states, the contraction and expansion of specific immune cell populations, and the remodeling of stromal architecture — all occurring on timescales of weeks to months. This temporal perspective is essential for biomarker development: a biomarker measured at diagnosis may become irrelevant after two cycles of therapy, and the predictive value of a given cellular composition depends on its dynamic trajectory, not merely its instantaneous state.
Spatial Context: From Dissociated Cells to Intact Tissue Architecture
Single-cell sequencing, for all its power, requires tissue dissociation — the physical destruction of the spatial relationships that organize cells into functional units. Spatial transcriptomics and spatial proteomics technologies address this limitation by profiling gene or protein expression in situ, preserving the anatomical context that determines cellular behavior. De Zuani and colleagues applied integrated scRNA-seq and spatial transcriptomics to NSCLC, demonstrating that the spatial proximity of specific cell types — for instance, the juxtaposition of exhausted T cells with PD-L1-expressing macrophages — carries prognostic information that cannot be recovered from dissociated cell analyses alone [PMID: 38782901]. Their work illustrates a fundamental principle: cells do not act in isolation, and the functional output of a cell is determined as much by its neighbors as by its intrinsic transcriptional state.
Liu and colleagues constructed a comprehensive atlas of immune heterogeneity in NSCLC by integrating data across multiple patients and tissue compartments, revealing spatial patterns of immune organization — from densely infiltrated "hot" regions to immune-excluded "cold" zones — within individual tumors [PMID: 40147443]. This intratumoral spatial heterogeneity has direct implications for biopsy-based biomarker assessment: a single core needle biopsy may sample an immune-hot region in one patient and an immune-cold region in the tumor next door, producing misleading biomarker readouts that reflect sampling artifact rather than true tumor biology. Tagore and colleagues extended the spatial paradigm to the metastatic setting, performing spatial molecular profiling of brain metastases from NSCLC and identifying distinct spatial niches with unique immune and stromal compositions [PMID: 40016452]. Brain metastases represent a particularly challenging clinical context where the blood-brain barrier, the specialized CNS immune environment, and the metabolic demands of the neural niche create selective pressures that are absent in the primary tumor — pressures whose effects are now becoming visible through spatial profiling technologies.
Computational Innovation: AI-Enabled Virtual Spatial Proteomics
The cost and technical complexity of spatial profiling technologies limit their application at population scale, creating an opportunity for computational methods that can infer spatial information from more accessible data modalities. Li and colleagues developed an AI-based platform for virtual spatial proteomics that predicts protein expression patterns from standard histopathology images, effectively generating spatial proteomic maps without the need for specialized spatial profiling assays [PMID: 41491099]. This approach, if validated at scale, could democratize spatial biomarker assessment by reducing it to a computational overlay on routine H&E-stained tissue sections — slides that are already available for virtually every diagnosed lung cancer patient. The implications are transformative: spatial biomarkers that currently require expensive, low-throughput assays could become universally accessible, enabling population-level studies of spatial immune organization and its relationship to treatment outcome.
The proliferation of single-cell and spatial platforms raises important questions about cross-platform comparability and reproducibility. Ozirmak Lermi and colleagues systematically compared multiple single-cell and spatial profiling platforms, evaluating concordance in cell type identification, gene detection sensitivity, and spatial resolution [PMID: 41006245]. Their analysis revealed substantial platform-dependent variation in sensitivity and specificity, with implications for meta-analysis and cross-study comparison. The field's reliance on platform-specific technical artifacts — dropout events in droplet-based scRNA-seq, spot size limitations in spatial transcriptomics — creates a reproducibility challenge that must be addressed through standardized benchmarking and harmonization protocols before single-cell and spatial data can be reliably integrated across studies and clinical sites.
Circulating Tumor DNA: From Detection to Ultra-Sensitive Monitoring
While tissue-based single-cell and spatial approaches provide unparalleled molecular resolution, they require invasive biopsies and capture only a single anatomical snapshot. Circulating tumor DNA (ctDNA) analysis offers a complementary, minimally invasive approach to real-time tumor monitoring. Abbosh and colleagues, working within the TRACERx consortium, demonstrated that ctDNA dynamics track tumor evolution and predict clinical relapse in early-stage NSCLC, establishing ctDNA as a tool for minimal residual disease detection [PMID: 37055640]. The clinical implication is profound: patients whose ctDNA becomes undetectable after surgery may be spared adjuvant chemotherapy, while those with persistent or rising ctDNA can be identified for early intensification of treatment. Black and colleagues pushed the sensitivity frontier further, developing ultrasensitive ctDNA detection methods capable of identifying tumor-derived DNA fragments at concentrations previously below the limit of detection [PMID: 41205598]. Ultrasensitive ctDNA monitoring has the potential to shift the therapeutic paradigm from reactive treatment of clinically evident recurrence to proactive intervention guided by molecular signals of minimal residual disease — a shift that demands new trial designs and new biomarker validation frameworks.
Multi-Modal Single-Cell Profiling: Metabolomics and Lipidomics
The most ambitious frontier in single-cell biology extends beyond transcriptomics to encompass metabolomic and lipidomic profiling at single-cell resolution. Li and colleagues developed methods for single-cell metabolic profiling, demonstrating that metabolic phenotypes vary dramatically across cells within a tumor and that metabolic heterogeneity predicts functional states — including drug sensitivity and immune evasion capacity — that are invisible to transcriptomic analysis alone [PMID: 31451693]. Wang and colleagues integrated scRNA-seq with lipidomic profiling, revealing coordinated changes in lipid metabolism and transcriptional programs that define distinct tumor cell subpopulations [PMID: 35108060]. Li and colleagues further advanced single-cell lipidomics, establishing technical frameworks for profiling lipid species at single-cell resolution and demonstrating the biological significance of lipid heterogeneity in tumor progression [PMID: 34001877]. These multi-modal approaches are still technically demanding and low-throughput, but they represent the logical endpoint of the resolution revolution: the ability to characterize every molecular layer of a cell — genome, transcriptome, proteome, metabolome, lipidome — simultaneously and in spatial context.
The Microbiome Question: An Important Negative Result
The intersection of microbiome research and lung cancer has generated considerable excitement, fueled by smaller studies suggesting that the intratumoral or airway microbiome may influence tumor biology, immune response, and clinical outcomes. McElderry and colleagues conducted the largest study to date on this question, profiling the microbiome in 940 never-smoker lung cancers — a cohort of extraordinary size and homogeneity that provides statistical power far exceeding any prior analysis [PMID: 41387456]. Their central finding is a critically important negative result: after rigorous quality control for contamination and batch effects, the study identified no clinically relevant associations between microbiome composition and lung cancer biology, contradicting the positive signals reported in smaller, less rigorously controlled studies. This finding does not exclude the possibility of microbiome-cancer interactions in specific clinical contexts — for example, in smokers with chronic airway inflammation, or in patients receiving antibiotics that disrupt commensal ecology — but it substantially deflates the hypothesis that the intratumoral microbiome is a major determinant of lung cancer phenotype in the general never-smoker population. The McElderry study is a salutary reminder that the history of cancer biology is littered with associations identified in underpowered studies that dissolve upon rigorous, large-scale replication, and that negative results, when they emerge from well-powered investigations, are as informative as positive ones.
Convergence: Toward Integrated Multi-Scale Tumor Models
The technologies surveyed in this chapter — single-cell transcriptomics, spatial profiling, ctDNA monitoring, single-cell metabolomics and lipidomics — are converging toward an integrated, multi-scale model of lung cancer biology that connects molecular events within individual cells to tissue-level spatial organization to organism-level circulating biomarkers. This convergence is not merely additive; the integration of data across scales and modalities creates emergent insights that are inaccessible to any single technology. The computational challenge is formidable: integrating sparse, noisy, and heterogeneously formatted data from platforms with different biases, resolutions, and dynamic ranges requires new statistical frameworks, new benchmarking standards, and new validation paradigms. But the scientific prize is equally formidable: a comprehensive, dynamic, multi-scale portrait of how lung cancers grow, evolve, resist therapy, and interact with the host immune system — a portrait that could transform both biological understanding and clinical decision-making.
 Infographic generated via NotebookLM from the chapter source material.
Infographic generated via NotebookLM from the chapter source material.
Implications for the Manuscript
This theme represents the technological vanguard of the corpus and should be positioned as the frontier toward which the field is moving. Key points for the manuscript include: (1) the foundational importance of single-cell atlases in redefining cellular heterogeneity beyond what bulk profiling could reveal; (2) the critical transition from dissociated-cell to spatial approaches that preserve tissue architecture and enable spatial biomarker discovery; (3) the potential of AI-enabled virtual spatial proteomics to democratize spatial profiling for clinical applications; (4) the complementarity of tissue-based and liquid biopsy-based approaches for multi-scale tumor monitoring; (5) the emergence of single-cell metabolomics and lipidomics as new dimensions of tumor characterization; and (6) the McElderry et al. finding as an important negative result that should temper enthusiasm for microbiome-cancer associations in the absence of large, rigorously controlled studies. Platform comparison studies should be cited as evidence that the field needs standardization before cross-study integration can be reliable. The 2 Tier A and 108 Tier B papers provide a robust evidence base for a forward-looking chapter that balances technological optimism with methodological rigor.