Environmental Exposures and Lung Cancer
"The air we breathe is not a passive medium; it is a pharmacological cocktail of particulates, gases, and radioactive decay products, each with its own dose-response curve for carcinogenesis."
 Infographic generated via NotebookLM from the chapter source material.
Infographic generated via NotebookLM from the chapter source material.
Landscape stats: 4,023 papers mapped to this theme (2 Tier A, 79 Tier B). The literature spans ambient air pollution, residential and occupational radon, asbestos, diesel exhaust, indoor biomass combustion, and emerging sensor technologies for exposure assessment.
Air Pollution as a Lung Cancer Promoter: The PM2.5 Paradigm
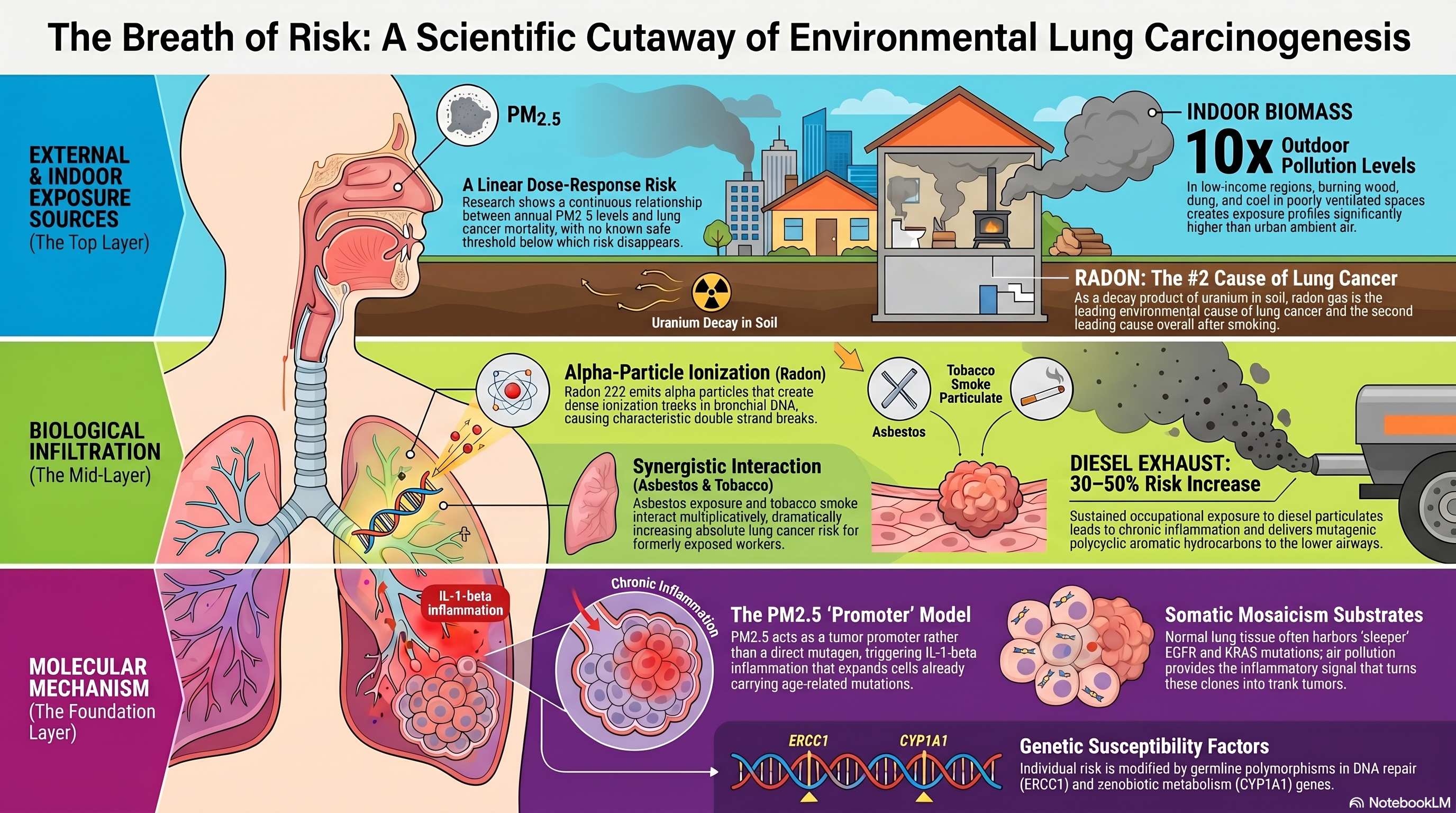
The relationship between fine particulate matter (PM2.5) and lung cancer has been studied for decades, but it was the mechanistic insights of the past several years that transformed this association from an epidemiological observation into a biologically coherent model. Pope and colleagues published one of the foundational studies linking long-term PM2.5 exposure to cardiopulmonary mortality, including lung cancer, in a large American Cancer Society cohort [PMID: 11879110]. Their analysis demonstrated a continuous, approximately linear dose-response relationship between annual average PM2.5 concentration and lung cancer mortality, with no apparent threshold below which risk disappeared entirely. This finding had profound public health implications because it suggested that even modest reductions in ambient PM2.5 could yield measurable reductions in lung cancer death, a prediction subsequently confirmed by natural experiments created by clean-air legislation in multiple countries.
Raaschou-Nielsen and colleagues extended the epidemiological evidence to European populations through an analysis of multiple prospective cohorts, confirming that ambient air pollution, specifically PM2.5 and nitrogen oxides, was associated with increased lung cancer incidence even at exposure levels below contemporary European regulatory limits [PMID: 23849838]. The European data were important because they demonstrated that the PM2.5-lung cancer relationship was not an artifact of the higher exposure levels found in some North American or Asian settings but persisted at concentrations encountered by hundreds of millions of people living in ostensibly well-regulated environments. The absence of a safe threshold was consistent with the Pope findings and reinforced the World Health Organization's decision to classify outdoor air pollution as a Group 1 carcinogen.
The mechanistic breakthrough came from Martinez-Ruiz and colleagues, who demonstrated that PM2.5 does not cause lung cancer through direct mutagenesis but rather acts as a tumor promoter by inducing inflammatory signaling in airway epithelial cells that already harbor pre-existing oncogenic mutations [PMID: 37046093]. Working within the framework of the TRACERx consortium, the investigators showed that normal lung tissue frequently contains clonal populations of cells carrying EGFR and KRAS mutations, the result of age-related somatic mosaicism, and that PM2.5 exposure triggers an interleukin-1-beta-mediated inflammatory response that promotes the expansion of these pre-malignant clones into frank tumors. This promoter model elegantly resolved a longstanding paradox: PM2.5 exposure is strongly associated with lung cancer incidence, yet air pollution does not leave a distinctive mutational signature in the tumor genome. The Martinez-Ruiz findings also provided a mechanistic link between air pollution and never-smoker lung cancer, because the pre-existing EGFR mutations that serve as substrates for PM2.5 promotion are precisely the driver mutations enriched in never-smoker tumors.
Murphy and colleagues provided a comprehensive review of environmental risk factors for lung cancer in never-smokers, contextualizing the PM2.5 promotion hypothesis within the broader landscape of non-tobacco carcinogens [PMID: 41114991]. Their synthesis emphasized that environmental exposures may interact with one another and with host genetic susceptibility in complex, non-additive ways, cautioning against the tendency to evaluate each exposure in isolation.
Indoor Air Pollution and Biomass Combustion
While ambient outdoor air pollution has received the most attention in high-income countries, indoor air pollution from biomass fuel combustion remains the dominant environmental lung cancer risk factor in low- and middle-income settings. A recent meta-analysis synthesized the evidence linking indoor air pollution from cooking and heating fuels, including wood, coal, dung, and crop residues, to lung cancer risk, finding a consistent elevation in risk among women who cooked with solid fuels in poorly ventilated spaces [PMID: 40950280]. The exposure profiles in these settings differ qualitatively from ambient PM2.5: indoor biomass combustion generates complex mixtures enriched in polycyclic aromatic hydrocarbons, volatile organic compounds, and ultrafine particles, with peak concentrations that can exceed outdoor levels by an order of magnitude. The epidemiological evidence for indoor pollution is particularly important for explaining the high rates of lung cancer among never-smoking women in rural China and South Asia, where biomass fuel use coincides with the highest female never-smoker lung cancer incidence globally.
Radon: The Invisible Domestic Carcinogen
Residential radon exposure represents the second leading cause of lung cancer after active smoking and the leading environmental cause in many countries. The biology of radon-induced carcinogenesis is well established: radon-222, a decay product of naturally occurring uranium-238 in soil and rock, emits alpha particles upon decay that cause dense ionization tracks in bronchial epithelial DNA, producing characteristic double-strand breaks and chromosomal aberrations. Radford provided early quantitative evidence from studies of underground miners exposed to high radon concentrations, demonstrating a clear dose-response relationship between cumulative radon exposure (measured in working-level months) and lung cancer mortality [PMID: 6325913]. The extrapolation of miner data to residential settings, where exposures are orders of magnitude lower but temporally prolonged, was a critical and contested step in radon risk assessment.
Pershagen and colleagues addressed this gap by conducting a case-control study of residential radon exposure and lung cancer in Sweden, using measured radon concentrations in subjects' homes to estimate cumulative exposure [PMID: 8264737]. Their analysis demonstrated a statistically significant dose-response relationship between residential radon and lung cancer risk, with the excess risk concentrated among subjects with the highest estimated exposures. Tomesek and colleagues contributed additional evidence from a Central European cohort, examining the spectrum of cancers associated with residential radon and confirming lung cancer as the primary malignancy associated with domestic radon exposure [PMID: 8096265].
The molecular signature of radon-induced carcinogenesis has been partially characterized. Vahakangas and colleagues examined p53 mutations in lung tumors from radon-exposed individuals and reported a distinctive pattern of transversion mutations, particularly G-to-T changes, consistent with the expected mutagenic consequences of alpha-particle radiation [PMID: 1347094]. While not entirely specific to radon (similar transversions are caused by benzo[a]pyrene in tobacco smoke), the p53 mutation spectrum provided molecular plausibility for the epidemiological association and suggested that radon-induced tumors might be distinguished from smoke-induced tumors by multi-gene mutation profiling. The practical challenge of radon mitigation has driven innovation in exposure assessment: Kholopo and colleagues described advances in radon sensor technology that enable continuous, real-time monitoring of indoor radon concentrations, facilitating both individual risk assessment and population-level surveillance [PMID: 38793821].
Asbestos: A Legacy Carcinogen with Persistent Burden
Asbestos exposure, though now regulated or banned in most high-income countries, continues to produce a substantial burden of lung cancer and mesothelioma due to the long latency period between exposure and disease manifestation. Camus and colleagues conducted a critical evaluation of the epidemiological evidence for chrysotile asbestos and lung cancer risk, examining cohort studies of workers exposed to varying fiber types and concentrations [PMID: 9603793]. Their analysis supported a dose-response relationship between cumulative asbestos exposure and lung cancer risk, though the magnitude of risk varied by fiber type (amphibole versus chrysotile) and by the co-occurrence of smoking, which multiplicatively increased lung cancer risk in asbestos-exposed individuals. The synergy between asbestos and tobacco smoke is one of the best-documented examples of multiplicative carcinogenic interaction and has implications for risk communication in formerly exposed populations where smoking cessation can substantially reduce absolute risk.
Diesel Exhaust and Occupational Exposures
Diesel engine exhaust, classified as a Group 1 carcinogen by IARC, represents an occupational hazard affecting millions of workers in transportation, mining, and construction. Lipsett and colleagues conducted a meta-analysis of epidemiological studies of diesel exhaust exposure and lung cancer risk, finding a consistent positive association across studies despite heterogeneity in exposure assessment methods [PMID: 10394308]. The meta-analytic estimate, indicating an approximately 30-50 percent increase in lung cancer risk among workers with sustained diesel exhaust exposure, was robust to sensitivity analyses and supported a causal interpretation. The biological mechanism is thought to involve both the mutagenic activity of polycyclic aromatic hydrocarbons adsorbed onto diesel particulate matter and the chronic inflammatory response triggered by particle deposition in the lower airways, paralleling the PM2.5 promotion model described by Martinez-Ruiz but with a more clearly mutagenic component.
Gene-Environment Interactions and Susceptibility
The observation that only a fraction of individuals with significant environmental exposures develop lung cancer has motivated investigations into gene-environment interactions. Germline polymorphisms in xenobiotic metabolism genes (CYP1A1, GSTM1, NAT2), DNA repair genes (ERCC1, XRCC1), and inflammatory mediator genes (IL-1-beta, TNF-alpha) have been associated with modified risk in the context of environmental carcinogen exposure, though individual effect sizes are small and replication has been inconsistent. The Martinez-Ruiz promoter model introduces a new framework for gene-environment interaction: if PM2.5 acts by promoting clones with pre-existing EGFR mutations, then the density of such clones in normal lung tissue, which itself may vary with age, sex, and inherited factors, becomes the critical determinant of susceptibility. This model predicts that individuals with higher somatic mutation burden in normal airway epithelium, a quantity now measurable by deep sequencing, will be at greater risk from a given level of PM2.5 exposure.
Implications for the Manuscript
The environmental exposures literature carries several implications for the manuscript. First, the Martinez-Ruiz PM2.5 promotion study should be featured prominently as a paradigm-shifting contribution that links air pollution to the molecular biology of lung carcinogenesis through a non-mutagenic, inflammation-mediated mechanism, and its connection to never-smoker EGFR-driven tumors should be explicitly drawn. Second, the radon literature, from Radford's miner studies through Pershagen's residential data to Kholopo's sensor innovations, provides a complete arc from hazard identification to molecular mechanism to mitigation technology that can be presented as a model for environmental cancer prevention. Third, the indoor pollution meta-analysis should be emphasized as a global health equity issue, because the populations most affected by biomass fuel exposure are precisely those with the least access to molecular diagnostics and targeted therapies. Fourth, the synergistic interaction between asbestos and tobacco smoke, documented by Camus, should be discussed as evidence that environmental and behavioral risk factors do not operate independently, reinforcing the need for integrated risk models. The manuscript should advocate for exposure assessment to be incorporated into the clinical evaluation of lung cancer patients, not merely as an epidemiological exercise but as a contributor to molecular subtyping and therapeutic decision-making.