Multi-Omics Integration Methods
"The challenge is no longer generating molecular data — it is making sense of the deluge, finding the signal that emerges only when disparate data types are brought into dialogue with one another."
 Infographic generated via NotebookLM from the chapter source material.
Infographic generated via NotebookLM from the chapter source material.
Literature base: 5,289 papers identified | 6 Tier A (landmark/highly cited) | 196 Tier B (significant contributions)
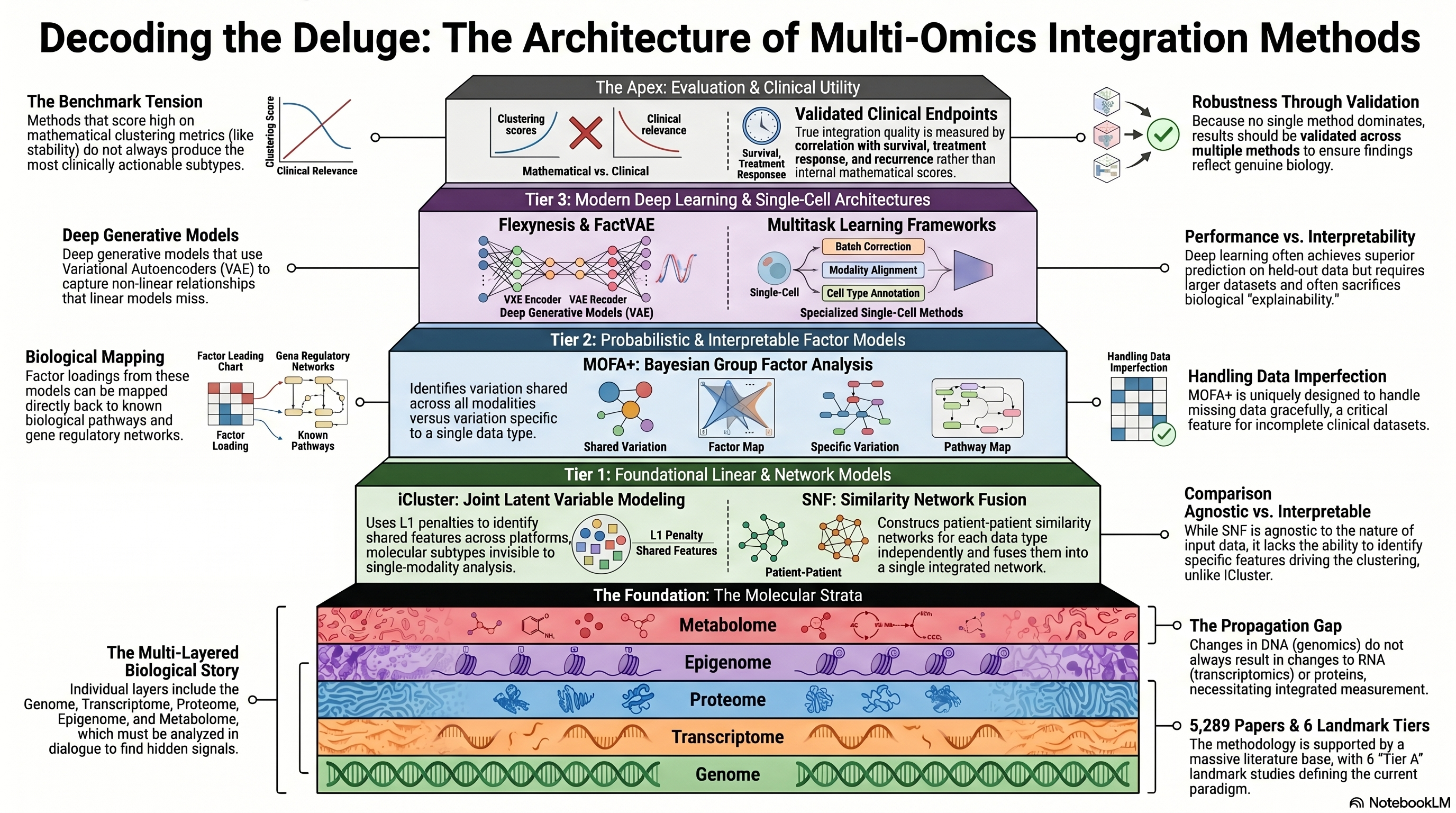
The proliferation of high-throughput molecular profiling technologies over the past two decades has created an embarrassment of riches: for any given tumor, it is now possible to measure the genome, transcriptome, proteome, epigenome, and metabolome with remarkable depth and resolution. Yet the fundamental insight driving the multi-omics integration field is that no single molecular layer tells the complete biological story. Genomic mutations may or may not alter transcript levels; transcriptomic changes may or may not propagate to protein abundance; and protein-level alterations may be invisible without measurement of post-translational modifications. The methods designed to integrate these complementary data streams represent one of the most active areas of computational biology, encompassing approaches that range from classical matrix factorization to sophisticated deep generative models. Understanding the strengths, limitations, and appropriate use cases of these methods is essential for interpreting the growing body of multi-omics studies in lung cancer and for designing the next generation of integrative analyses.
Foundational Approaches: Matrix Factorization and Network Fusion
The intellectual origins of multi-omics integration can be traced to methods that sought to identify shared latent structure across multiple data matrices. iCluster, introduced by Shen, Olshen, and Ladanyi, was among the first principled approaches to jointly cluster tumors using data from multiple molecular platforms [PMID: 19759197]. The method employs a joint latent variable model in which each data type is modeled as a linear function of a shared set of latent factors, with an L1 penalty to enforce sparsity and identify the features most relevant to cluster definition. iCluster and its successors established the paradigm of discovering molecular subtypes that are invisible to any single data modality but emerge when multiple layers are analyzed in concert — a paradigm that has been applied extensively in TCGA pan-cancer analyses and remains influential today.
A fundamentally different philosophy underlies Similarity Network Fusion (SNF), developed by Wang and colleagues, which constructs patient similarity networks from each data type independently and then fuses them into a single integrated network using an iterative diffusion process [PMID: 24464287]. SNF sidesteps the challenge of modeling the joint distribution of heterogeneous data types by operating entirely in the space of patient-patient similarities, making it agnostic to the specific nature of the input data. The method demonstrated strong performance in identifying cancer subtypes with clinical relevance and has been widely adopted for its conceptual simplicity and scalability. However, SNF treats all data types as equally informative and does not provide a mechanism for identifying which features within each data type drive the observed clustering — a limitation that motivated subsequent work on interpretable integration methods.
Modern Factor Models: MOFA+ and the Quest for Interpretability
The Multi-Omics Factor Analysis framework, extended as MOFA+ by Argelaguet and colleagues, represents a significant advance in the direction of interpretable, probabilistic multi-omics integration [PMID: 32393329]. MOFA+ employs a Bayesian group factor analysis model in which each data modality contributes to a set of shared and modality-specific latent factors, with automatic relevance determination to identify which factors capture variation that is shared across data types versus variation that is specific to a single modality. The method can handle missing data gracefully — a critical practical advantage, since clinical multi-omics datasets are rarely complete — and provides interpretable factor loadings that can be mapped back to biological pathways and processes. MOFA+ has been applied to both bulk and single-cell multi-omics data, and its output can be integrated with downstream analyses such as differential expression, trajectory inference, and gene regulatory network reconstruction. The framework has become a standard reference point for benchmarking new integration methods, reflecting both its theoretical elegance and practical utility.
The extension of these ideas to single-cell multi-omics has introduced additional challenges related to data sparsity, high dimensionality, and the need to account for cellular heterogeneity within samples. Hu and colleagues conducted a comprehensive benchmarking study of single-cell multi-omics integration methods, evaluating their performance across metrics including cluster preservation, batch effect correction, and biological signal retention [PMID: 39322753]. Their analysis revealed that no single method dominated across all evaluation criteria, and that the optimal choice of integration strategy depends heavily on the specific characteristics of the dataset and the biological question being addressed. This finding has important implications for the lung cancer field, where single-cell multi-omics is increasingly used to dissect tumor heterogeneity and microenvironment composition but where the sensitivity of results to methodological choices is not always appreciated.
Benchmarking and Evaluation: What Makes a Good Integration?
A persistent challenge in the multi-omics integration field has been the lack of consensus on how to evaluate integration quality. Pierre-Jean and colleagues undertook a systematic benchmarking of multi-omics clustering methods, comparing the performance of iCluster, SNF, and several other approaches on both simulated and real cancer datasets [PMID: 31792509]. Their study revealed substantial variation in performance across methods and datasets, with no single approach emerging as universally superior. Notably, method performance was highly sensitive to the degree of concordance between data types: methods that assume strong shared structure tended to perform well when data types were highly correlated but poorly when inter-modal associations were weak. Chauvel and colleagues extended this benchmarking effort with a rigorous evaluation framework that assessed not only clustering accuracy but also the biological interpretability and reproducibility of the identified subtypes [PMID: 31220206]. Their work highlighted a tension that pervades the field: methods that achieve the best quantitative clustering metrics do not always produce the most biologically meaningful or clinically actionable subtypes, and vice versa.
These benchmarking studies collectively underscore several practical lessons for researchers working in lung cancer multi-omics. First, the choice of integration method should be guided by the specific biological question: unsupervised subtyping, supervised biomarker discovery, and network inference each favor different methodological approaches. Second, results should be validated across multiple methods to assess robustness, as findings that are method-specific are unlikely to reflect genuine biological signal. Third, the evaluation of integration quality should incorporate clinical endpoints — survival, treatment response, recurrence — rather than relying solely on internal metrics such as silhouette scores or cluster stability.
Deep Learning Approaches: From Autoencoders to Foundation Models
The application of deep learning to multi-omics integration has accelerated dramatically in recent years, driven by the promise of learning complex, non-linear relationships between data modalities that linear methods cannot capture. Athaya and colleagues provided a comprehensive review of multimodal deep learning approaches for biomedical data integration, cataloguing architectures that range from simple concatenation-based fully connected networks to sophisticated attention-based models and variational autoencoders [PMID: 37651607]. They noted that while deep learning methods often achieve superior predictive performance on held-out data, they typically sacrifice interpretability and require substantially larger training datasets than traditional statistical approaches — constraints that are particularly relevant in the oncology setting, where sample sizes are often limited and biological interpretability is essential for clinical translation.
Flexynesis, developed by Uyar and colleagues, represents a recent effort to bridge the gap between deep learning performance and practical usability in multi-omics integration [PMID: 40940333]. The framework provides a flexible architecture that can accommodate varying numbers of input modalities and supports multiple downstream tasks, including classification, regression, and survival prediction, within a unified modeling framework. The tool is designed with the bioinformatics end-user in mind, providing automated hyperparameter optimization and interpretability modules that extract feature importance scores from trained models. Similarly, FactVAE, introduced by Wang and colleagues, employs a variational autoencoder architecture with a factorized latent space that disentangles shared and modality-specific variation — conceptually similar to MOFA+ but within a deep generative modeling framework capable of capturing non-linear relationships [PMID: 40211981].
The application of deep learning to single-cell multi-omics integration has produced several notable methods. Liu and colleagues developed a multitask learning framework for single-cell data integration that simultaneously performs batch correction, modality alignment, and cell type annotation, leveraging the complementary information across tasks to improve performance on each [PMID: 41083898]. Mowgli, introduced by Huizing and colleagues, takes a different approach by combining optimal transport theory with non-negative matrix factorization to integrate single-cell multi-omics data, providing both dimensionality reduction and joint embedding in a mathematically principled framework [PMID: 38001063]. These single-cell methods are particularly relevant for lung cancer research, where dissecting the cellular composition of the tumor microenvironment requires the joint analysis of multiple molecular modalities at single-cell resolution.
Reviews and the State of the Art
Several recent reviews have attempted to synthesize the rapidly expanding landscape of multi-omics integration methods and provide guidance for practitioners. Baiao and colleagues offered a comprehensive overview of computational approaches for multi-omics data integration, organizing methods along axes of supervised versus unsupervised learning, early versus late fusion strategies, and traditional statistical versus deep learning approaches [PMID: 40748323]. Their analysis emphasized that the field is transitioning from method development to method application, with the primary bottleneck shifting from the availability of integration tools to the availability of well-curated, multi-modal datasets with clinical annotation. This observation resonates strongly with the lung cancer field, where large-scale multi-omics datasets such as those from TCGA and CPTAC exist but are often analyzed in isolation rather than as integrated wholes.
Where Consensus Exists and Where the Field Disagrees
The multi-omics methods community has converged on several key principles. There is broad agreement that no single molecular data type is sufficient to capture the full complexity of cancer biology and that integrative approaches are essential. The importance of benchmarking on standardized datasets with clinically relevant endpoints has been widely acknowledged, even if not universally practiced. The complementary strengths of linear factor models (interpretability, robustness with small sample sizes) and deep learning approaches (flexibility, capacity for non-linear relationships) are generally recognized, with most investigators accepting that the appropriate method depends on the dataset and the question.
Disagreements remain, however, on several fronts. The relative merits of early fusion (combining raw features before analysis) versus late fusion (analyzing each data type separately and combining results) versus intermediate fusion (learning joint latent representations) continue to be debated, with no definitive resolution emerging from benchmarking studies. The question of whether deep learning methods offer genuine biological advantages over simpler approaches for multi-omics integration in oncology — as opposed to merely better performance on leaderboard metrics — remains contentious. The degree to which single-cell multi-omics integration methods, developed primarily on model systems and healthy tissues, are appropriate for the technically challenging and often degraded specimens typical of clinical oncology is also uncertain.
MOVICS Dominance and the Feature Selection Philosophy Divide
While the preceding sections describe a rich and diverse methodological landscape, the actual practice of multi-omics integration in lung cancer tells a strikingly different story. A systematic census of 25 multi-omics lung cancer studies reveals that 18 of 25 — a commanding majority — employ MOVICS (Multi-Omics Integrative Clustering System), a consensus-based framework published by Lu et al. (2020) in Bioinformatics. MOVICS aggregates results across 10 distinct clustering algorithms, including iCluster, SNF, and several others discussed in this chapter, to produce a consensus subtype assignment. Its appeal lies in the promise of robustness: by requiring agreement across methods, MOVICS ostensibly protects against the method-specific artifacts highlighted by the benchmarking literature. Yet this near-universal adoption has created a methodological monoculture in the lung cancer field, where the diversity of available integration tools — factor models, network fusion, deep learning — is acknowledged in review articles but rarely exercised in practice.
Within the MOVICS-dominated landscape, the single biggest determinant of downstream results is not the integration algorithm itself but the upstream feature selection strategy. Of the 18 MOVICS-based lung cancer studies, 13 employ Cox regression-based supervised feature selection, in which genes are retained only if they show a statistically significant association with overall survival. This survival-driven filtering consistently yields binary (two-subtype) classifications that map onto high-risk versus low-risk groupings. By contrast, only 3 of 18 studies use variance-based unsupervised feature selection, which retains the most variable features regardless of their relationship to clinical outcome. These variance-based studies consistently identify 4 to 5 subtypes with distinct biological programs. The implication is profound: the number and nature of the subtypes reported in a given study are largely predetermined by the feature selection philosophy, not discovered de novo by the integration algorithm. This methodological choice — supervised versus unsupervised feature selection — deserves far greater scrutiny than it has received, as it fundamentally shapes the biological conclusions drawn from otherwise similar analytical pipelines.
It is also notable that DIABLO, the supervised multi-block PLS-DA method that has demonstrated success in breast cancer multi-omics integration, has seen zero applications in lung cancer. Despite its ability to identify features that jointly discriminate known classes across multiple data modalities, no lung cancer study has tested whether DIABLO's supervised integration approach could complement or outperform the consensus clustering paradigm. This absence represents both a gap and an opportunity: the lung cancer field's methodological convergence on a single framework means that alternative approaches remain entirely unexplored, and findings that might emerge from genuinely different analytical philosophies are simply missing from the literature.
Critical Gaps
Several critical gaps impede the effective application of multi-omics integration methods to lung cancer research. First, most methods have been developed and evaluated on bulk tissue data and may not adequately handle the spatial and temporal heterogeneity that characterizes real tumors. The integration of spatial multi-omics data — which preserves tissue architecture — requires methods that can model spatial dependencies alongside molecular correlations, a capability that most existing frameworks lack. Second, the integration of clinical data (demographics, treatment history, imaging) with molecular data remains methodologically underdeveloped, despite the obvious clinical importance of considering the full patient context. Third, there is a pressing need for standardized preprocessing pipelines and quality control metrics for multi-omics data, as variations in data processing can have outsized effects on integration results. Fourth, the computational cost and expertise required to implement state-of-the-art integration methods remain barriers to adoption by the broader lung cancer research community, highlighting the need for user-friendly tools and comprehensive documentation.
Implications for the Manuscript
This chapter provides the methodological foundation for the review, establishing the analytical toolkit that will be applied to lung cancer in the subsequent chapter on multi-omics applications. The progression from iCluster and SNF through MOFA+ to deep generative models mirrors the increasing sophistication of the biological questions being asked in lung cancer research, from simple subtype discovery to mechanistic understanding of therapy resistance and immune evasion. The benchmarking literature reviewed here provides the critical context for evaluating the integration methods employed in lung cancer studies: when a study reports multi-omics subtypes, the reader must consider whether those subtypes are robust to methodological choice, whether they have been validated in independent cohorts, and whether they add clinical value beyond existing biomarkers. The gap between method development and clinical application identified in this chapter will be a recurring theme throughout the manuscript, and the recommendations for best practices in multi-omics integration will inform the review's concluding guidance for the field.