Epigenetics and Chromatin Remodeling in Lung Cancer
"The genome is the script, but the epigenome is the director, and in lung cancer the director has gone rogue in ways we are only beginning to catalog."
 Infographic generated via NotebookLM from the chapter source material.
Infographic generated via NotebookLM from the chapter source material.
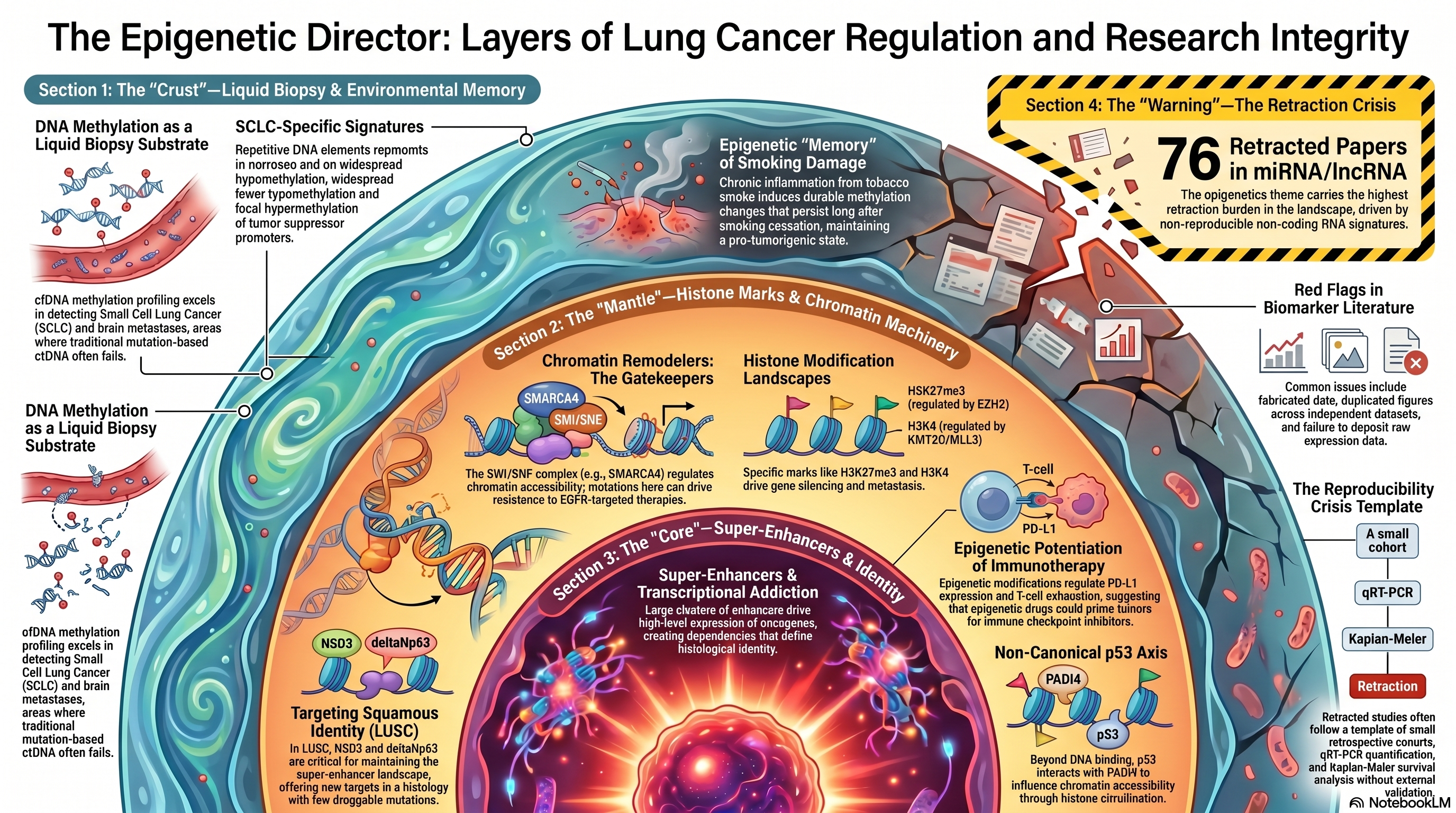
Landscape stats: 5,902 papers mapped to this theme (0 Tier A, 91 Tier B, 76 retracted). This is the largest theme by paper count and, notably, carries the highest retraction burden of any theme in our landscape analysis. The concentration of retractions in miRNA/lncRNA prognostic signature studies constitutes a reproducibility crisis within the subfield that warrants explicit discussion.
The Retraction Crisis: A Field Contaminated by Non-Reproducible Signatures
Before engaging with the substantive epigenetics literature, the manuscript must confront an uncomfortable finding: of the 5,902 papers mapped to this theme, 76 have been retracted, a rate that far exceeds any other theme in our landscape analysis. The retractions are not randomly distributed across the epigenetics literature but are heavily concentrated in a specific genre of study: papers reporting miRNA or long non-coding RNA (lncRNA) expression signatures as prognostic or diagnostic biomarkers. The typical retracted paper follows a recognizable template: a small retrospective cohort, qRT-PCR quantification of a panel of non-coding RNAs, Kaplan-Meier survival analysis with a median-split dichotomization, and claims of independent prognostic value in multivariate models. Investigations by journal editors and post-publication reviewers have identified fabricated data, duplicated figures across ostensibly independent datasets, implausible statistical results, and failure to deposit raw data as recurring features of retracted studies.
The implications of this retraction cluster extend beyond the individual papers withdrawn. The retracted studies have been cited thousands of times collectively, and their findings have been incorporated into meta-analyses, systematic reviews, and narrative reviews that remain in the literature. Several of the retracted non-coding RNA biomarkers have been proposed as candidates for clinical validation studies, meaning that downstream research investment may have been misdirected. The retraction pattern also reflects a structural vulnerability in the epigenetics biomarker field: the ease of generating small-cohort expression data, combined with the high-dimensional nature of non-coding RNA profiling (which facilitates overfitting), creates conditions that are fertile for both honest false discoveries and deliberate fabrication. The manuscript should flag this retraction burden as a cautionary finding and recommend that future non-coding RNA biomarker studies adhere to REMARK guidelines, deposit raw expression data in public repositories, and undergo independent external validation before being considered for clinical translation.
DNA Methylation as a Liquid Biopsy Substrate
Despite the reproducibility concerns in the non-coding RNA space, several areas of epigenetics research have yielded robust, clinically translatable findings. Cell-free DNA (cfDNA) methylation profiling has emerged as one of the most promising liquid biopsy approaches for lung cancer detection and monitoring. Heeke and colleagues demonstrated the utility of cfDNA methylation analysis for detecting small cell lung cancer (SCLC), a histology that has proven resistant to conventional ctDNA mutation-based approaches due to the relative scarcity of recurrent point mutations in SCLC genomes [PMID: 38278149]. Their methylation-based assay achieved high sensitivity and specificity for distinguishing SCLC from NSCLC and from non-cancer controls, exploiting the distinctive epigenomic landscape of SCLC, which is characterized by widespread hypomethylation of repetitive elements and focal hypermethylation of tumor suppressor gene promoters. The clinical value of a SCLC-specific liquid biopsy is considerable, given that SCLC is frequently diagnosed at advanced stages when tissue biopsy is challenging and that histological transformation from NSCLC to SCLC is an increasingly recognized mechanism of treatment resistance.
Zuccato and colleagues applied cfDNA methylation profiling to detect brain metastases in lung cancer patients, demonstrating that methylation patterns in circulating tumor DNA could identify patients with central nervous system involvement with clinically useful accuracy [PMID: 39379704]. Brain metastases represent a major source of morbidity and mortality in advanced NSCLC, and the blood-brain barrier complicates both detection and monitoring. A peripheral blood-based assay capable of flagging CNS disease would represent a meaningful advance in surveillance and early intervention. The biological basis for this approach rests on the observation that metastatic tumors to the brain acquire organ-specific epigenomic alterations, including methylation changes at genes involved in neural adhesion and blood-brain barrier interaction, that distinguish them from the primary tumor and from metastases to other sites.
Inflammation, Methylation, and the Tumor Microenvironment
The relationship between chronic inflammation and epigenetic reprogramming is central to lung carcinogenesis, particularly in the context of environmental exposures and smoking-related damage. Wielscher and colleagues conducted an epigenome-wide association study linking peripheral blood DNA methylation patterns to inflammatory biomarkers and lung cancer risk, identifying methylation changes at immune-regulatory loci that mediated the association between systemic inflammation and subsequent lung cancer development [PMID: 35504910]. Their findings supported a model in which chronic inflammation induces durable epigenetic changes in immune and epithelial cells that persist even after the inflammatory stimulus is removed, creating a "memory" of prior exposure that predisposes to malignant transformation. This epigenetic memory concept has implications for understanding why lung cancer risk remains elevated for decades after smoking cessation: the methylation changes induced by tobacco smoke may be incompletely reversible, maintaining a pro-tumorigenic epigenetic state long after the mutagen is no longer present.
Weng and colleagues reviewed the intersection of epigenetic mechanisms and immune regulation in lung cancer, synthesizing evidence that epigenetic modifications in both tumor cells and immune cells shape the tumor microenvironment and influence response to immunotherapy [PMID: 33846331]. Their analysis highlighted the role of DNA methylation and histone modifications in regulating PD-L1 expression, antigen presentation machinery, and T-cell exhaustion programs, suggesting that epigenetic therapies could potentiate immune checkpoint inhibitor efficacy.
Histone Modifications and Chromatin Remodeling
The histone modification landscape in lung cancer has been characterized by a series of studies identifying specific marks and their regulatory enzymes as drivers of tumor biology. Kondo and colleagues demonstrated that the polycomb repressive mark H3K27me3, deposited by the EZH2 methyltransferase complex, was a critical regulator of gene silencing in lung cancer, with widespread gains of H3K27me3 at tumor suppressor loci contributing to the malignant phenotype [PMID: 18488029]. This finding placed EZH2 inhibition on the therapeutic agenda for lung cancer, although subsequent clinical experience has demonstrated that EZH2 inhibitors as monotherapy have modest activity in solid tumors, likely because the functional consequences of H3K27me3 loss depend on the broader chromatin context.
The enzymes responsible for writing, reading, and erasing histone modifications have emerged as frequent targets of somatic mutation in lung cancer. Na and colleagues characterized the role of KMT2C (MLL3), a histone H3 lysine 4 methyltransferase, in SCLC metastasis, demonstrating that loss-of-function mutations in KMT2C disrupted enhancer activation at genes required for epithelial identity, facilitating the transition to a more motile, invasive phenotype [PMID: 35449309]. Ju and colleagues investigated NatD, an N-terminal acetyltransferase that modifies histone H4, and showed that NatD activity contributed to lung cancer cell proliferation by modulating chromatin accessibility at cell-cycle genes [PMID: 29030587]. These studies collectively illustrate the diversity of histone-modifying enzymes implicated in lung cancer and the challenge of prioritizing therapeutic targets within this crowded landscape.
Super-Enhancers and Transcriptional Addiction
The concept of super-enhancers, large clusters of enhancer elements that drive high-level expression of lineage-defining and oncogenic genes, has been particularly productive in lung cancer research. Alam and colleagues demonstrated that KMT2D (MLL4), a histone methyltransferase frequently mutated in lung cancer, was required for the maintenance of super-enhancer landscapes, and that KMT2D loss remodeled the super-enhancer repertoire in ways that activated oncogenic transcriptional programs [PMID: 32243837]. Yuan and colleagues identified NSD3, a histone methyltransferase amplified in lung squamous cell carcinoma (LUSC), as a driver of super-enhancer formation at loci critical for squamous cell identity and proliferation [PMID: 33536620]. The NSD3 findings were notable because LUSC has relatively few druggable genomic alterations compared with adenocarcinoma, and NSD3 amplification identified a potentially targetable epigenetic dependency in a histology with significant unmet therapeutic need.
Napoli and colleagues characterized the role of deltaNp63, a transcription factor overexpressed in squamous lung cancer, in establishing and maintaining enhancer landscapes that define squamous cell identity [PMID: 35105868]. Their work demonstrated that deltaNp63 occupied super-enhancers at squamous lineage genes and that its depletion led to enhancer decommissioning and loss of squamous identity, suggesting that the transcriptional programs maintaining histological identity are themselves epigenetically regulated and potentially druggable. Huang and colleagues described a role for circular RNAs (circRNAs) in modulating super-enhancer activity, adding a non-coding RNA dimension to the super-enhancer regulatory circuit [PMID: 40512546].
Chromatin Remodeling Complexes and Therapeutic Resistance
The SWI/SNF chromatin remodeling complex has emerged as a critical node in lung cancer biology, both as a target of frequent somatic mutation and as a determinant of therapeutic response. de Miguel and colleagues demonstrated that mutations in SWI/SNF complex subunits, including SMARCA4 and ARID1A, conferred resistance to tyrosine kinase inhibitors (TKIs) in EGFR-mutant NSCLC by altering chromatin accessibility at genes involved in bypass signaling pathways [PMID: 37541244]. This finding was clinically important because SWI/SNF mutations are present in a substantial minority of EGFR-mutant tumors and could potentially serve as predictive biomarkers for TKI resistance. The mechanistic link between chromatin remodeling and drug resistance extended the concept of resistance beyond the traditional framework of secondary mutations in the drug target, positioning epigenetic plasticity as a distinct and therapeutically addressable resistance mechanism.
p53, Epigenetic Cross-Talk, and Non-Canonical Regulatory Axes
The tumor suppressor p53, the most frequently mutated gene in lung cancer, exerts effects that extend beyond its well-characterized transcriptional activation program into the epigenetic domain. Tanikawa and colleagues identified a direct physical and functional interaction between p53 and PADI4, a peptidylarginine deiminase that converts arginine residues to citrulline on histone tails, altering chromatin structure and gene expression [PMID: 22334079]. The p53-PADI4 axis represented a non-canonical mechanism by which p53 could influence gene expression: rather than binding to promoter elements and recruiting transcriptional machinery, p53 could modulate chromatin accessibility through histone citrullination, a post-translational modification whose biological significance is still being elucidated. Osada and colleagues characterized the role of TGF-beta signaling, another pathway frequently dysregulated in lung cancer, in mediating epigenetic changes through induction of DNA methyltransferases and histone-modifying enzymes [PMID: 11719467]. The convergence of p53, TGF-beta, and epigenetic regulatory machinery suggests that the major tumor-suppressive pathways in lung cancer operate in part through chromatin-level mechanisms, a perspective that reframes therapeutic targeting of these pathways.
Implications for the Manuscript
The epigenetics theme generates several key messages for the manuscript. First, and most distinctively, the 76 retractions concentrated in miRNA/lncRNA prognostic signature studies should be discussed as a manuscript-worthy finding in its own right. The retraction pattern reveals structural vulnerabilities in the non-coding RNA biomarker field, including small sample sizes, lack of independent validation, and inadequate data sharing, that should be highlighted as a cautionary tale and accompanied by specific recommendations for raising the evidentiary bar. Second, cfDNA methylation-based liquid biopsies, exemplified by the Heeke SCLC study and the Zuccato brain metastasis study, represent a clinically mature application of epigenetic biology that should be positioned as complementary to mutation-based ctDNA approaches. Third, the super-enhancer and chromatin remodeling literature identifies actionable epigenetic dependencies, particularly in LUSC (NSD3, deltaNp63) and in TKI-resistant EGFR-mutant NSCLC (SWI/SNF), that should be discussed as therapeutic opportunities. Fourth, the manuscript should integrate the epigenetic dimension of the tumor microenvironment, drawing on the Wielscher and Weng studies, to argue that epigenetic therapies may have immunomodulatory effects that make them rational combination partners for immune checkpoint inhibitors. The combination of robust translational findings and the retraction crisis makes this theme both one of the most promising and one of the most cautionary in the lung cancer landscape.