Molecular Heterogeneity in Lung Cancer
"The more we learn about the molecular underpinnings of lung cancer, the more we appreciate that no two tumors — and perhaps no two cells within a single tumor — are truly alike."
 Infographic generated via NotebookLM from the chapter source material.
Infographic generated via NotebookLM from the chapter source material.
Literature base: 6,812 papers identified | 14 Tier A (landmark/highly cited) | 444 Tier B (significant contributions)
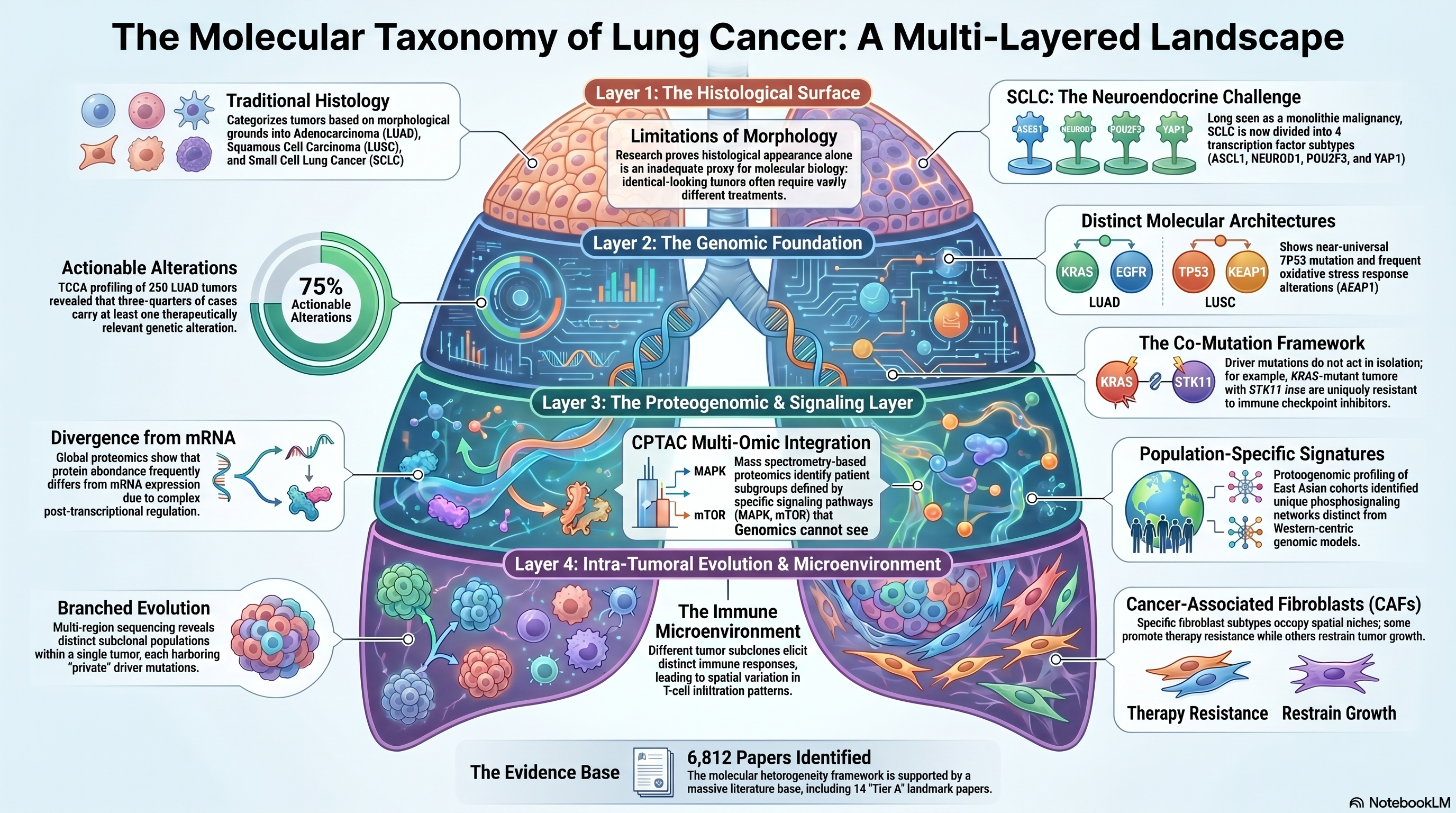
The recognition that lung cancer is not a single disease but a constellation of molecularly distinct entities has fundamentally reshaped how researchers and clinicians approach this malignancy. Over the past two decades, the field has migrated from purely histological classification schemes — distinguishing adenocarcinoma from squamous cell carcinoma from small cell lung cancer on morphological grounds alone — to a molecular taxonomy that stratifies tumors by driver mutations, transcriptomic programs, proteomic states, and epigenetic configurations. This paradigm shift has been driven by large-scale genomic profiling efforts, most notably those conducted under the umbrella of The Cancer Genome Atlas (TCGA), and has yielded actionable therapeutic insights that were inconceivable when lung cancer was treated as a monolithic disease. Yet even as the molecular landscape has been charted with increasing resolution, the sheer complexity and heterogeneity of these tumors continues to outpace our ability to translate molecular knowledge into durable clinical benefit for all patients.
The Genomic Foundation: TCGA and Large-Scale Profiling
The modern molecular portrait of lung adenocarcinoma (LUAD) was established by the TCGA Research Network, whose comprehensive genomic characterization of 230 tumors revealed an intricate landscape of somatic alterations spanning 18 statistically recurrent mutated genes [PMID: 25079552]. This seminal study delineated distinct molecular subtypes defined by mutations in TP53, KRAS, EGFR, and STK11, among others, and demonstrated that LUAD harbors a higher mutational burden than many other solid tumors, reflecting its strong association with tobacco carcinogenesis. Critically, the study identified that approximately 75% of LUAD tumors carried at least one therapeutically relevant alteration, establishing the rationale for precision oncology approaches that have since transformed the treatment landscape. In parallel, the TCGA effort on lung squamous cell carcinoma (LUSC) characterized 178 tumors and identified a strikingly different molecular architecture, with near-universal TP53 mutation, frequent alterations in the oxidative stress response pathway through KEAP1 and NFE2L2, and recurrent amplification of SOX2 and PIK3CA [PMID: 22960745]. The contrast between these two major histological subtypes underscored a fundamental lesson: histological appearance alone is an inadequate proxy for molecular biology, and therapeutic strategies must be grounded in genomic context rather than morphological classification.
The molecular characterization of small cell lung cancer (SCLC) lagged behind its non-small cell counterparts, in part because of the rarity of surgical specimens in a disease typically diagnosed at advanced stages. George and colleagues addressed this gap through comprehensive genomic profiling of 110 SCLC cases, confirming the near-universal inactivation of TP53 and RB1 and identifying recurrent mutations in chromatin-modifying genes such as CREBBP, EP300, and MLL [PMID: 26168399]. This work established that SCLC, long regarded as a relatively homogeneous neuroendocrine malignancy, possesses its own brand of molecular diversity. Building on these genomic foundations, Rudin and colleagues proposed a transcription factor-based classification of SCLC into four subtypes defined by differential expression of ASCL1, NEUROD1, POU2F3, and YAP1, each associated with distinct therapeutic vulnerabilities [PMID: 30926931]. This framework has since become the standard for SCLC subtyping and has guided the development of subtype-specific therapeutic strategies, including DLL3-targeting agents for ASCL1-high tumors. The practical validation of this classification scheme through immunohistochemistry demonstrated that these subtypes can be identified using routinely available clinical pathology methods, broadening the potential for clinical implementation [PMID: 33011388].
Co-Mutations and the Complexity of Driver Context
The early genomic studies established which genes are recurrently altered in lung cancer, but a more nuanced understanding has emerged from the recognition that driver mutations rarely act in isolation. Skoulidis and colleagues systematically characterized the co-mutation landscape of KRAS-mutant lung adenocarcinoma, revealing that concurrent alterations in STK11 and KEAP1 define biologically and clinically distinct subgroups with profoundly different prognoses and therapeutic responses [PMID: 31406302]. KRAS-mutant tumors with co-occurring STK11 loss, for instance, exhibit a uniquely immunosuppressive microenvironment characterized by reduced T-cell infiltration and diminished response to immune checkpoint inhibitors, whereas KRAS/TP53 co-mutant tumors tend to harbor higher mutational burden and greater immunogenicity. This co-mutation framework has had immediate clinical implications: the development and clinical testing of sotorasib for KRAS G12C-mutant NSCLC demonstrated that even within a molecularly defined population, co-mutation context modulates treatment benefit, with STK11 and KEAP1 co-mutations conferring resistance [PMID: 40437272]. Thomas and colleagues further reinforced the importance of molecular context by demonstrating how integrative genomic and clinical data can refine treatment selection beyond single-gene biomarkers, arguing for a more holistic assessment of the molecular landscape when making therapeutic decisions [PMID: 25963091].
The Proteomic and Proteogenomic Layer
While genomic characterization provided the foundational map of lung cancer heterogeneity, it became increasingly apparent that transcript and protein-level measurements capture biological variation not visible through DNA sequencing alone. The Clinical Proteomic Tumor Analysis Consortium (CPTAC) brought mass spectrometry-based proteomics to bear on the problem, generating comprehensive proteomic and phosphoproteomic profiles of LUAD tumors [PMID: 32649874]. Gillette and colleagues demonstrated that proteomic data could identify patient subgroups with distinct biology that were not apparent from genomic data alone, including subgroups defined by differential activation of signaling pathways such as MAPK and mTOR. Simultaneously, Xu and colleagues applied global proteomics to a large LUAD cohort and showed that protein abundance frequently diverges from mRNA expression, with post-transcriptional regulation playing a major role in shaping the functional state of tumors [PMID: 32649877]. The importance of integrating proteomics with genomics was further emphasized by Chen and colleagues, who profiled a cohort of East Asian lung adenocarcinomas enriched for EGFR-mutant tumors and identified proteogenomic features unique to this population, including distinct phosphosignaling networks not captured by Western-centric cohorts [PMID: 32649875]. These findings highlight the population-specific dimension of molecular heterogeneity and underscore the need for inclusive, multi-ethnic molecular profiling efforts.
The proteogenomic approach has been extended to SCLC, where Liu and colleagues integrated whole-genome sequencing, transcriptomics, and proteomics to characterize the molecular architecture of this aggressive disease at unprecedented resolution [PMID: 38181741]. Their analysis revealed that SCLC subtypes defined by transcription factor expression correspond to distinct proteomic and metabolic programs, and identified potential therapeutic targets — including druggable kinases — that were not apparent from genomic data alone. More recently, Song and colleagues performed deep proteogenomic profiling of NSCLC and identified molecular subtypes with prognostic and predictive significance that cut across traditional histological boundaries [PMID: 39580524]. These studies collectively demonstrate that proteogenomics adds an essential biological layer to the characterization of lung cancer heterogeneity and is likely to become increasingly important as therapeutic strategies target protein-level vulnerabilities.
Tumor Evolution and Intra-Tumoral Heterogeneity
Beyond the diversity observed across patients, a growing body of work has documented the remarkable molecular heterogeneity that exists within individual tumors. Martinez-Ruiz and colleagues performed multi-region whole-genome sequencing and transcriptomic profiling of NSCLC tumors to map the evolutionary trajectories of these cancers, revealing extensive branched evolution with distinct subclonal populations harboring private driver alterations [PMID: 37046093]. Their analysis demonstrated that the immune microenvironment co-evolves with the tumor, with different tumor subclones eliciting distinct immune responses and contributing to spatial variation in immune infiltration patterns. This intra-tumoral heterogeneity has profound implications for biomarker assessment, as single-biopsy sampling may fail to capture the full molecular diversity of a tumor and could lead to incomplete or misleading biomarker results.
The tumor microenvironment represents another axis of heterogeneity that is increasingly recognized as a critical determinant of clinical behavior. Hanley and colleagues performed single-cell and spatial transcriptomic profiling of lung tumors to characterize cancer-associated fibroblast (CAF) populations and demonstrated that distinct fibroblast subtypes occupy specific spatial niches within the tumor and exert opposing effects on tumor immunity and progression [PMID: 36720863]. Some CAF populations promote immunosuppression and therapy resistance, while others appear to restrain tumor growth, highlighting the inadequacy of simplistic models that treat the stroma as a homogeneous entity. These findings add yet another dimension to the heterogeneity challenge and suggest that effective therapeutic strategies may need to account for stromal biology alongside tumor cell-intrinsic features.
Rare Subtypes and the Long Tail of Molecular Diversity
While the major histological subtypes — LUAD, LUSC, and SCLC — have received the most intensive molecular characterization, lung cancer encompasses a long tail of rare histological variants whose molecular biology remains poorly understood. Harada and colleagues undertook a systematic molecular characterization of rare lung cancer subtypes, including large cell neuroendocrine carcinoma, adenosquamous carcinoma, and sarcomatoid variants, revealing that these tumors harbor distinct genomic and transcriptomic profiles that do not fit neatly into existing classification schemes [PMID: 36806787]. Their work demonstrated that some rare subtypes share molecular features with more common histologies — for instance, a subset of large cell neuroendocrine carcinomas resembles SCLC at the transcriptomic level — while others represent genuinely distinct molecular entities. The clinical implications are significant: patients with rare subtypes are typically excluded from major clinical trials and treated empirically, often with regimens designed for more common histologies, an approach that may be inappropriate given their distinct molecular biology.
Where Consensus Exists and Where the Field Disagrees
Several areas of broad consensus have emerged from the molecular characterization of lung cancer. There is near-universal agreement that NSCLC and SCLC are fundamentally different diseases at the molecular level and should be approached with distinct therapeutic strategies. Within NSCLC, the importance of identifying actionable driver mutations — EGFR, ALK, ROS1, BRAF V600E, KRAS G12C, and others — is well established and has been codified in clinical guidelines worldwide. The co-mutation framework for KRAS-mutant LUAD has gained widespread acceptance as a biologically and clinically meaningful stratification tool.
However, significant disagreements persist. The optimal molecular classification of SCLC remains debated: while the four-subtype model proposed by Rudin has been influential, some investigators argue that SCLC exists along a continuum of neuroendocrine differentiation rather than falling into discrete categories, and the clinical utility of subtype-specific therapeutic strategies remains unproven in randomized trials. The extent to which proteomic subtypes add actionable information beyond what is captured by genomic profiling is also contested, with skeptics arguing that the technical complexity and cost of mass spectrometry-based proteomics limit its clinical scalability. Furthermore, the appropriate level of spatial and temporal sampling needed to capture intra-tumoral heterogeneity in routine clinical practice remains unresolved, with pragmatic constraints of tissue availability clashing with the biological imperative for comprehensive sampling.
Critical Gaps
Several critical gaps remain in our understanding of molecular heterogeneity in lung cancer. First, the molecular basis of de novo and acquired resistance to targeted therapies and immunotherapies is incompletely understood, particularly the contribution of pre-existing subclonal heterogeneity to treatment failure. Second, the integration of proteomic, metabolomic, and epigenomic data with genomic profiles remains technically challenging and has not yet been achieved at scale in clinical settings. Third, the molecular characterization of lung cancer in underrepresented populations — including African American, Hispanic, and South Asian patients — is woefully incomplete, limiting the generalizability of current molecular taxonomies. Fourth, the tumor microenvironment, while increasingly recognized as a critical determinant of clinical behavior, has not been incorporated into routine molecular classification schemes in a clinically actionable manner. Finally, the rare histological subtypes of lung cancer remain molecularly undercharacterized, leaving a significant fraction of patients without evidence-based, molecularly guided treatment options.
Implications for the Manuscript
This chapter establishes molecular heterogeneity as the foundational challenge that motivates the entire review. The progression from single-gene biomarkers to multi-omic molecular taxonomies provides the logical scaffold for subsequent chapters on multi-omics integration methods and AI/ML approaches: these technologies are needed precisely because the molecular complexity of lung cancer exceeds what can be captured by any single data modality. The co-mutation framework and proteogenomic findings will be directly referenced in later chapters as examples of biological insights that require integrative analytical approaches. The discussion of intra-tumoral heterogeneity and tumor evolution sets the stage for spatial multi-omics methods covered in the applications chapter. The critical gaps identified here — particularly the need for population-inclusive profiling and clinically scalable integration strategies — will be revisited as unifying themes in the synthesis and future directions sections of the manuscript.