Multi-Omics Applications in Lung Cancer
"Methods without applications are academic exercises; applications without rigorous methods are sand castles. The studies that advance the field are those that bring both together in the service of genuine clinical need."
 Infographic generated via NotebookLM from the chapter source material.
Infographic generated via NotebookLM from the chapter source material.
Literature base: 2,648 papers identified | 1 Tier A (landmark/highly cited) | 127 Tier B (significant contributions)
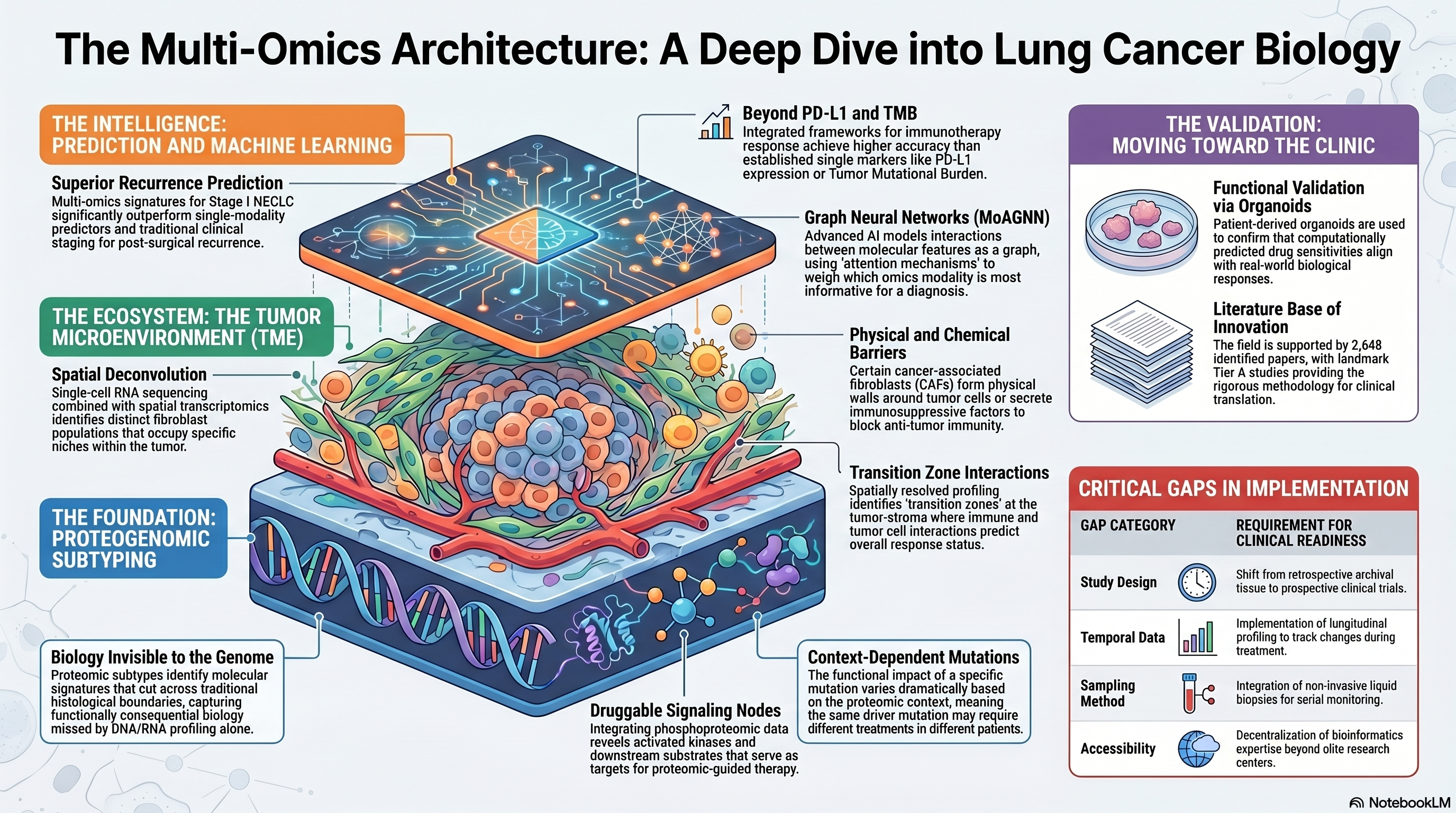
The translation of multi-omics integration methods from the computational arena to lung cancer biology represents one of the most consequential developments in thoracic oncology research over the past five years. While the preceding chapter surveyed the methodological landscape in abstract terms, this chapter examines how those methods have been deployed to answer specific biological and clinical questions in lung cancer: What molecular subtypes exist beyond traditional histological categories? How does the tumor microenvironment shape disease trajectory and treatment response? Can multi-omics signatures predict which patients will benefit from specific therapies, or which early-stage cancers will recur after surgery? The studies reviewed here demonstrate that multi-omics integration is no longer a proof-of-concept exercise but is beginning to generate clinically meaningful insights — while also revealing the substantial gap that remains between research discovery and routine clinical implementation.
Proteogenomic Subtyping: Beyond the Genome
Among the most impactful applications of multi-omics integration in lung cancer has been the discovery of proteogenomic subtypes that capture biology invisible to genomic or transcriptomic profiling alone. Lehtio and colleagues performed deep proteogenomic characterization of a large NSCLC cohort, integrating genomic, transcriptomic, and proteomic data to identify molecular subtypes with distinct clinical trajectories and therapeutic vulnerabilities [PMID: 34870237]. Their analysis revealed that proteomic subtypes cut across traditional histological boundaries, with some adenocarcinomas and squamous cell carcinomas sharing proteomic features that suggest convergent biology despite divergent genomic origins. Critically, the proteogenomic subtypes provided prognostic information that was independent of — and complementary to — established clinical and genomic risk factors, suggesting that proteomic data captures aspects of tumor biology that are functionally consequential but not reflected in the genome.
Song and colleagues extended this line of investigation through comprehensive proteogenomic profiling of NSCLC, employing an integrative analytical framework that combined mass spectrometry-based proteomics with whole-exome sequencing and RNA sequencing [PMID: 39580524]. Their study identified molecular subtypes defined by differential activation of metabolic, immune, and cell cycle programs at the protein level, and demonstrated that these subtypes were associated with distinct patterns of response to chemotherapy and immunotherapy. The integration of phosphoproteomic data allowed the identification of druggable signaling nodes — activated kinases and their downstream substrates — that were not detectable from genomic data alone, providing a template for proteomic-guided therapeutic selection. Su and colleagues further advanced the proteogenomic characterization of lung cancer by focusing on the relationship between genomic alterations and their downstream proteomic consequences, revealing that the functional impact of specific mutations varies dramatically depending on the proteomic context in which they occur [PMID: 40069142]. This finding has profound implications for precision oncology, suggesting that the same driver mutation may confer different therapeutic vulnerabilities in different patients depending on the broader molecular state of their tumors.
The Tumor Microenvironment Through a Multi-Omics Lens
Perhaps no area of lung cancer biology has benefited more from multi-omics integration than the characterization of the tumor microenvironment (TME). The recognition that tumors are ecosystems — comprising not only malignant cells but also fibroblasts, immune cells, endothelial cells, and other stromal components — has driven the adoption of technologies and analytical approaches that can deconvolve the contributions of these distinct cell populations. Hanley and colleagues employed single-cell RNA sequencing combined with spatial transcriptomics to characterize cancer-associated fibroblast diversity in lung tumors, identifying functionally distinct fibroblast populations that occupy specific spatial niches and differentially influence immune cell recruitment and anti-tumor immunity [PMID: 36720863]. Their work demonstrated that certain fibroblast subtypes form physical barriers between immune cells and tumor cells, while others actively secrete immunosuppressive factors, providing a mechanistic explanation for the spatially variable immune infiltration patterns observed in many lung cancers.
The advent of spatial multi-omics technologies has opened new windows into the architecture of the TME. Aung and colleagues applied spatially resolved transcriptomic and proteomic profiling to map molecular signatures across the spatial dimensions of lung tumors, revealing that immune activation, metabolic reprogramming, and signaling pathway activity vary dramatically across different regions of the same tumor [PMID: 41073787]. Their spatial analysis identified transition zones at the tumor-stroma interface where immune and tumor cells interact most intensively, and demonstrated that the molecular characteristics of these transition zones are predictive of overall immune response status. Chen and colleagues built on these spatial approaches to characterize intra-tumoral spatial heterogeneity at multi-omic resolution, showing that spatially distinct tumor regions harbor different molecular subtypes and evolutionary lineages, and that this spatial heterogeneity has implications for the accuracy of biomarker assessment from single biopsies [PMID: 39983726].
Clinical Prediction: Recurrence, Immunotherapy, and Beyond
The ultimate test of any molecular characterization effort is whether it improves clinical decision-making. Several recent studies have applied multi-omics integration to the clinically urgent problem of predicting outcomes and guiding treatment selection in lung cancer. Wang and colleagues addressed the challenge of predicting recurrence in stage I NSCLC — a population for which current risk stratification is inadequate, leading to both overtreatment and undertreatment — by developing a multi-omics signature that integrates genomic, transcriptomic, and clinical features to identify patients at high risk of post-surgical recurrence [PMID: 39929832]. Their model significantly outperformed single-modality predictors and existing clinical staging, suggesting that multi-omics integration may enable more precise adjuvant therapy decisions in early-stage disease.
The prediction of immunotherapy response represents another high-value application of multi-omics integration. Wang and colleagues developed a multi-omics framework for predicting response to immune checkpoint inhibitors in lung squamous cell carcinoma, integrating genomic mutation data, transcriptomic immune signatures, and proteomic markers to achieve superior predictive accuracy compared to established biomarkers such as PD-L1 expression and tumor mutational burden [PMID: 39762577]. Their analysis revealed that no single biomarker adequately captures the complexity of the tumor-immune interaction, and that the integration of complementary molecular measurements is necessary to stratify patients into clinically meaningful response categories. This finding aligns with the broader recognition in the immunotherapy field that the determinants of treatment benefit are multifactorial and cannot be reduced to a single molecular marker.
Graph Neural Networks and Machine Learning Integration
The application of advanced machine learning architectures to multi-omics data has introduced new analytical paradigms for lung cancer research. Lin and colleagues developed MoAGNN, a multi-omics attention graph neural network that models interactions between molecular features as a graph and learns to weight the contributions of different omics modalities to prediction tasks through an attention mechanism [PMID: 41554048]. The graph-based architecture naturally captures the network structure of biological systems — where genes, proteins, and metabolites interact in complex regulatory networks — and the attention mechanism provides interpretability by revealing which molecular features and interactions are most informative for a given prediction. Applied to lung cancer subtyping and outcome prediction, MoAGNN demonstrated superior performance compared to both traditional statistical methods and standard deep learning architectures.
Zhang and colleagues took a complementary approach, using machine learning methods to identify robust molecular subtypes of lung cancer from multi-omics data and to characterize the biological programs that distinguish these subtypes [PMID: 39669580]. Their analysis employed ensemble approaches that aggregated results across multiple integration methods and multiple resampling iterations, addressing the robustness concerns raised by benchmarking studies and producing subtypes that were reproducible across independent cohorts. The emphasis on methodological rigor and reproducibility in their analytical framework provides a valuable model for the field, where the temptation to overfit complex models to small datasets is ever-present.
Functional Validation and Model Systems
While computational multi-omics integration has generated a wealth of hypotheses about lung cancer biology, the functional validation of these hypotheses remains a critical bottleneck. Fukushima and colleagues addressed this challenge by combining multi-omics profiling with patient-derived organoid models, allowing computational predictions to be tested in physiologically relevant experimental systems [PMID: 40307487]. Their study demonstrated that organoid drug sensitivity patterns correlate with multi-omics molecular subtypes, providing functional evidence that the subtypes identified through computational integration reflect genuine biological differences with therapeutic implications. This integration of computational and experimental approaches represents the gold standard for multi-omics studies and should be encouraged as a prerequisite for claims of clinical relevance.
The relevance of multi-omics approaches extends beyond lung cancer to inform pan-cancer methodology. Migliozzi and colleagues applied spatial multi-omics integration to glioblastoma and developed analytical frameworks for combining spatially resolved transcriptomics with proteomic and metabolomic data that are broadly applicable across tumor types [PMID: 36732634]. While the specific biological findings pertain to brain tumors, the methodological innovations — including algorithms for spatially aware data integration and frameworks for quantifying spatial heterogeneity — are directly transferable to lung cancer research, where spatial multi-omics is an emerging frontier.
Where Consensus Exists and Where the Field Disagrees
There is growing consensus that multi-omics integration provides biological and clinical information beyond what any single data modality can offer. The value of proteogenomic approaches, in particular, has been convincingly demonstrated by multiple independent groups, and the identification of proteomic subtypes that cut across histological boundaries represents a genuine advance in our understanding of lung cancer biology. The importance of the tumor microenvironment as a determinant of clinical behavior is also broadly accepted, and multi-omics approaches have become the standard tool for its characterization.
Disagreements center on the clinical readiness of multi-omics approaches. Critics argue that the technical complexity, cost, and turnaround time of multi-omics profiling render it impractical for routine clinical use, and that simpler biomarkers — even if less biologically comprehensive — may be more appropriate for clinical implementation. The question of whether proteomic subtypes provide sufficient added clinical value over genomic and transcriptomic profiling alone to justify the additional complexity and cost remains actively debated. The generalizability of multi-omics signatures across populations, platforms, and clinical settings has also not been adequately demonstrated, with most studies relying on retrospective cohorts from a small number of academic medical centers. Furthermore, the optimal study design for clinically validating multi-omics biomarkers — including the appropriate endpoints, sample sizes, and validation strategies — has not been standardized.
The Census View: Cohort Homogeneity, Feature Selection Blind Spots, and Missing Modalities
A systematic census of 25 multi-omics lung cancer studies reveals a striking dependence on a single data source: 22 of 25 studies (88%) use TCGA as their primary discovery cohort. Only two studies employ institutional cohorts with original multi-omics profiling — Zhang et al. (2025, Nature Communications), who characterized 122 stage I NSCLC patients, and Zhong et al. (2025, Molecular Cancer), who profiled 101 early-stage LUAD cases. The remaining study uses a GEO-derived dataset. This near-total reliance on TCGA means that the field's conclusions about multi-omics subtypes, prognostic signatures, and therapeutic vulnerabilities are effectively derived from a single retrospective cohort collected over a decade ago, with all the attendant limitations in clinical annotation, treatment heterogeneity, and demographic representation. The generalizability of these findings to contemporary patient populations receiving modern therapies remains largely untested.
A subtler but equally consequential problem concerns the role of clinical features in multi-omics analyses. Across the 25 studies, clinical variables such as age, tumor stage, smoking history, and sex are universally treated as validation endpoints — variables against which molecular subtypes are tested for association — rather than as input features that compete with molecular data for predictive value. No study in the census tests whether its multi-omics signature adds incremental prognostic or predictive value over clinical features alone. This omission is critical because clinical staging already provides substantial prognostic information, and the bar for clinical utility is not whether a molecular signature predicts outcome in isolation but whether it improves upon what clinicians already know. Furthermore, the dominant Cox-based supervised feature selection approach introduces a related distortion: by collapsing subtypes into binary risk groups, it merges biologically distinct mechanisms under a single "high-risk" label. Zou et al. (2022) demonstrated that unsupervised biological clustering reveals four subtypes with distinct drug sensitivities — including immune-excluded and metabolically dysfunctional phenotypes that carry different therapeutic implications but would be collapsed into a single high-risk category under binary risk stratification. The loss of this therapeutic information represents a direct cost of the prognosis-first analytical philosophy.
Finally, the census reveals striking gaps in data modality coverage. Zero studies integrate metabolomics data, despite growing evidence that metabolic reprogramming is a defining feature of lung cancer subtypes. Zero studies incorporate spatial omics at the multi-omics integration level, despite the spatial technologies discussed earlier in this chapter. Proteomics integration is minimal: only 2 of 25 studies use RPPA (reverse-phase protein array) data, and none leverage the deep mass spectrometry-based proteomics available through CPTAC. The multi-omics label, as currently practiced in the lung cancer literature, overwhelmingly denotes the combination of mRNA expression, DNA methylation, somatic mutations, and copy number alterations — a configuration that, while informative, leaves substantial biological dimensions unmeasured and unintegrated.
Critical Gaps
Several critical gaps limit the clinical translation of multi-omics findings in lung cancer. First, the vast majority of existing multi-omics studies are retrospective, relying on archival tissue from surgical resections, and their findings have not been validated in prospective clinical trials. Second, the integration of longitudinal multi-omics data — tracking molecular changes over the course of disease and treatment — remains technically challenging and has been attempted in only a handful of studies. Third, the application of multi-omics integration to non-invasive liquid biopsy samples, which could enable serial monitoring without repeated tissue sampling, is in its infancy. Fourth, the computational infrastructure and bioinformatics expertise required to perform multi-omics integration remain concentrated in a small number of well-resourced research centers, creating a significant barrier to the democratization of these approaches. Fifth, the functional validation of computationally derived multi-omics subtypes and biomarkers through experimental model systems such as organoids and genetically engineered mouse models remains the exception rather than the rule.
Implications for the Manuscript
This chapter provides the clinical and biological anchor for the review, demonstrating that multi-omics integration has moved beyond methodological demonstration to address genuine clinical questions in lung cancer. The proteogenomic subtyping studies establish a direct link between the methods reviewed in the preceding chapter and the clinical heterogeneity discussed in the molecular heterogeneity chapter, showing that integration methods can discover biology that matters for patients. The spatial multi-omics studies preview the emerging frontier of tissue-architecture-aware molecular analysis, which will be further discussed in the context of AI/ML approaches in the following chapter. The critical gaps identified here — particularly the need for prospective validation, longitudinal profiling, and liquid biopsy integration — will form key components of the review's future directions section and will be positioned as priority areas for the field.